Hypervalent Iodine Reagents in High Valent Transition Metal Chemistry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

BI(III) Initiated Cyclization Reactions and Iodonium Fragmentation Kinetics
W&M ScholarWorks Dissertations, Theses, and Masters Projects Theses, Dissertations, & Master Projects 2005 BI(III) Initiated Cyclization Reactions and Iodonium Fragmentation Kinetics Yajing Lian College of William & Mary - Arts & Sciences Follow this and additional works at: https://scholarworks.wm.edu/etd Part of the Inorganic Chemistry Commons Recommended Citation Lian, Yajing, "BI(III) Initiated Cyclization Reactions and Iodonium Fragmentation Kinetics" (2005). Dissertations, Theses, and Masters Projects. Paper 1539626842. https://dx.doi.org/doi:10.21220/s2-zj7g-fp23 This Thesis is brought to you for free and open access by the Theses, Dissertations, & Master Projects at W&M ScholarWorks. It has been accepted for inclusion in Dissertations, Theses, and Masters Projects by an authorized administrator of W&M ScholarWorks. For more information, please contact [email protected]. BI(III) INITIATED CYCLIZATION REACTIONS AND IODONIUM FRAGMENTATION KINETICS A Thesis Presented to The Faculty of the Department of Chemistry The College of William and Mary in Virginia In Partial Fulfillment Of the Requirements for the Degree of Master of Science by Yajing Lian 2005 APPROVAL SHEET This thesis is submitted in partial fulfillment of The requirements for the degree of Master of Science A Yajing Lian Approved by the Committee, December 20, 2005 Robert J. Hinkle, Chair Christopher Jl A] Robert D. Pike TABLE OF CONTENTS Page Acknowledgements VI List of Tables Vll List of Schemes V lll List of Figures IX Abstract XI Introduction Chapter I Secondary -

Design, Synthesis and Applications of Novel Hypervalent Iodine Reagents
Design, Synthesis and Applications of Novel Hypervalent Iodine Reagents A Thesis Submitted to Cardiff University in Fulfilment of the Requirements for the Degree of Doctor of Philosophy by Jihan Qurban PhD Thesis July 2019 Cardiff University Jihan Qurban PhD Thesis 2019 __________________________________________________________________________________ DECLARATION This work has not been submitted in substance for any other degree or award at this or any other university or place of learning, nor is being submitted concurrently in candidature for any degree or other award. Signed …………………………… (Jihan Qurban) Date ………………….… STATEMENT 1 This thesis is being submitted in partial fulfillment of the requirements for the degree of ……………………… (insert MCh, MD, MPhil, PhD etc, as appropriate) Signed …………………………… (Jihan Qurban) Date ………………….… STATEMENT 2 This thesis is the result of my own independent work/investigation, except where otherwise stated, and the thesis has not been edited by a third party beyond what is permitted by Cardiff University’s Policy on the Use of Third-Party Editors by Research Degree Students. Other sources are acknowledged by explicit references. The views expressed are my own. Signed …………………………… (Jihan Qurban) Date ………………….… STATEMENT 3 I hereby give consent for my thesis, if accepted, to be available online in the University’s Open Access repository and for inter-library loan, and for the title and summary to be made available to outside organisations. Signed …………………………… (Jihan Qurban) Date ………………….… STATEMENT 4: PREVIOUSLY APPROVED BAR ON ACCESS I hereby give consent for my thesis, if accepted, to be available online in the University’s Open Access repository and for inter-library loans after expiry of a bar on access previously approved by the Academic Standards & Quality Committee. -

Oxidative Addition & Reductive Elimination
3/1/2021 Oxidative Addition & Reductive Elimination Robert H. Crabtree: Pages 159 – 182 and 235 - 266 1 Oxidative Addition Change in Example Group configuration • Oxidative addition is a key step in many transition-metal catalyzed reactions 10 8 I III • Basic reaction: d d Cu Cu 11 X X Pd0 PdII 10 LnM + LnM Pt0 PtII 10 Y Y d8 d6 PdII PdIV 10 • The new M-X and M-Y bonds are formed using the electron pair of the X-Y PtII PtIV 10 bond and one metal-centered lone pair IrI IrIII 9 RhI RhIII 9 • The metal goes up in oxidation state (+2), X-Y formally gets reduced to X-, Y- d6 d4 ReI ReIII 7 • The ease of addition (or elimination) can be tuned by the electronic and 0 II steric properties of the ancillary ligands Mo Mo 6 • OA favored by strongly e-donating L d4 d2 MoII MoIV 6 • The most common applications involve: a) Late transition metals (platinum metals: Ru, Rh, Pd, Ir, Pt) - not d7 d6 2CoII 2CoIII 9 too sensitive to O2 and H2O; routinely used in organic synthesis b) C-Halogen, H-H or Si-H bonds d4 d3 2CrII 2CrIII 6 • Common for transition metals, rare for main-group metals but: Grignard reagents! 2 1 3/1/2021 Oxidative addition – concerted mechanism Concerted addition - mostly with non-polar X-Y bonds X X X LnM + LnM LnM Y Y Y A Cr(CO)5: • H2, silanes, alkanes, ... coordinatively Cr(PMe3)5: • Arene C-H bonds more reactive than alkane C-H bonds unsaturated, but metal- centered lone pairs not phosphines are better • H-H, C-H strong bond, but M-H and M-C bonds can be very available donors, weaker acceptors full oxidative -

A Metal-Free Approach to Biaryl Compounds: Carbon-Carbon Bond Formation from Diaryliodonium Salts and Aryl Triolborates
Portland State University PDXScholar Dissertations and Theses Dissertations and Theses Winter 4-3-2015 A Metal-Free Approach to Biaryl Compounds: Carbon-Carbon Bond Formation from Diaryliodonium Salts and Aryl Triolborates Kuruppu Lilanthi Jayatissa Portland State University Follow this and additional works at: https://pdxscholar.library.pdx.edu/open_access_etds Part of the Catalysis and Reaction Engineering Commons Let us know how access to this document benefits ou.y Recommended Citation Jayatissa, Kuruppu Lilanthi, "A Metal-Free Approach to Biaryl Compounds: Carbon-Carbon Bond Formation from Diaryliodonium Salts and Aryl Triolborates" (2015). Dissertations and Theses. Paper 2229. https://doi.org/10.15760/etd.2226 This Thesis is brought to you for free and open access. It has been accepted for inclusion in Dissertations and Theses by an authorized administrator of PDXScholar. Please contact us if we can make this document more accessible: [email protected]. A Metal-Free Approach to Biaryl Compounds: Carbon-Carbon Bond Formation from Diaryliodonium Salts and Aryl Triolborates. by Kuruppu Lilanthi Jayatissa A thesis submitted in partial fulfillment of the requirement for the degree of Master of Science in Chemistry Thesis Committee: David Stuart, Chair Robert Strongin Theresa McCormick Portland State University 2015 Abstract Biaryl moieties are important structural motifs in many industries, including pharmaceutical, agrochemical, energy and technology. The development of novel and efficient methods to synthesize these carbon-carbon bonds is at the forefront of synthetic methodology. Since Ullmann’s first report of stoichiometric Cu-mediated homo-coupling of aryl halides, there has been a dramatic evolution in transition metal catalyzed biaryl cross-coupling reactions. -

Metathetical Redox Reaction of (Diacetoxyiodo)Arenes and Iodoarenes
Article Metathetical Redox Reaction of (Diacetoxyiodo)arenes and Iodoarenes Antoine Jobin-Des Lauriers and Claude Y. Legault * Received: 25 November 2015 ; Accepted: 15 December 2015 ; Published: 17 December 2015 Academic Editors: Wesley Moran and Arantxa Rodríguez Centre in Green Chemistry and Catalysis, Department of Chemistry, University of Sherbrooke, 2500 boul. de l’Université, Sherbrooke, QC J1K 2R1, Canada; [email protected] * Correspondence: [email protected]; Tel./Fax: +1-819-821-8017 Abstract: The oxidation of iodoarenes is central to the field of hypervalent iodine chemistry. It was found that the metathetical redox reaction between (diacetoxyiodo)arenes and iodoarenes is possible in the presence of a catalytic amount of Lewis acid. This discovery opens a new strategy to access (diacetoxyiodo)arenes. A computational study is provided to rationalize the results observed. Keywords: hypervalent iodine; (diacetoxyiodo)arenes; metathesis 1. Introduction In the recent years, the development of hypervalent iodine-mediated synthetic transformations has been receiving growing attention [1–5]. This is not surprising considering the importance of improving oxidative reactions and reducing their environmental impact. Hypervalent iodine reagents are a great alternative to toxic heavy metals often used to effect similar transformations [6–10]. Among the plethora of iodine(III) compounds [11], it is undeniable that the (diacetoxyiodo)arenes remain the most common and used ones [12]. They are usually stable solids and can serve as precursors to numerous other iodanes, such as other (diacyloxyiodo)arenes and [hydroxy(tosyloxy) iodo]arenes [13,14]. Access to (diacetoxyiodo)arenes is, of course, usually accomplished by the oxidation of the corresponding iodoarene substrate. -

SELECTIVE C–H and ALKENE FUNCTIONALIZATION VIA ORGANIC PHOTOREDOX CATALYSIS Nicholas Paul Ralph Onuska a Dissertation Submitte
SELECTIVE C–H AND ALKENE FUNCTIONALIZATION VIA ORGANIC PHOTOREDOX CATALYSIS Nicholas Paul Ralph Onuska A dissertation submitted to the faculty at the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Chemistry Chapel Hill 2021 Approved by: David Nicewicz Erik Alexanian Jeffrey Aubé Alexander Miller Andrew Moran © 2021 Nicholas Paul Ralph Onuska ALL RIGHTS RESERVED ii ABSTRACT Nicholas Paul Ralph Onuska: Selective C–H and Alkene Functionalization Via Organic Photoredox Catalysis (Under the direction of David A. Nicewicz) Photoinduced electron transfer (PET) can be defined as the promotion of an electron transfer reaction through absorption of a photon by a reactive species in solution. Photoredox catalysis has leveraged previous knowledge of PET processes to enable a wide variety of high value chemical transformations. Most early work in the field of photoredox catalysis centered around the use of visible-light absorbing transition metal catalyst systems based on ruthenium or iridium polypyridyl scaffolds, organic redox chromophores offer a more sustainable, accessible, and versatile alternative to these complexes. Organic photoredox catalysts are often less expensive, easier to prepare, and have expanded redox windows relative to many known transition-metal photoredox catalysts, enabling the generation of a wider variety of single electron reduced/oxidized species. This ultimately translates into the development of previously undiscovered modes of reactivity. Herein is described the development and mechanistic study of a several C–H and alkene functionalization reactions facilitated by visible light-absorbing acridinium salts, as well as the discovery and characterization of a neutral organic radical which possess one of the most negative excited state oxidation potentials observed to date. -

Chapter 9 Hydrogen
Chapter 9 Hydrogen Diborane, B2 H6• is the simplest member of a large class of compounds, the electron-deficient boron hydrid Like all boron hydrides, it has a positive standard free energy of formation, and so cannot be prepared direct from boron and hydrogen. The bridge B-H bonds are longer and weaker than the tenninal B-H bonds (13: vs. 1.19 A). 89.1 Reactions of hydrogen compounds? (a) Ca(s) + H2Cg) ~ CaH2Cs). This is the reaction of an active s-me: with hydrogen, which is the way that saline metal hydrides are prepared. (b) NH3(g) + BF)(g) ~ H)N-BF)(g). This is the reaction of a Lewis base and a Lewis acid. The product I Lewis acid-base complex. (c) LiOH(s) + H2(g) ~ NR. Although dihydrogen can behave as an oxidant (e.g., with Li to form LiH) or reductant (e.g., with O2 to form H20), it does not behave as a Br0nsted or Lewis acid or base. It does not r with strong bases, like LiOH, or with strong acids. 89.2 A procedure for making Et3MeSn? A possible procedure is as follows: ~ 2Et)SnH + 2Na 2Na+Et3Sn- + H2 Ja'Et3Sn- + CH3Br ~ Et3MeSn + NaBr 9.1 Where does Hydrogen fit in the periodic chart? (a) Hydrogen in group 1? Hydrogen has one vale electron like the group 1 metals and is stable as W, especially in aqueous media. The other group 1 m have one valence electron and are quite stable as ~ cations in solution and in the solid state as simple 1 salts. -

Enantioselective Iodine(I/III) Catalysis in Organic Synthesis, Which Has Been Published in Final Form At
"This is the peer reviewed version of the following article: Enantioselective Iodine(I/III) Catalysis in Organic Synthesis, which has been published in final form at https://doi.org/10.1002/adsc.201800521 . This article may be used for non-commercial purposes in accordance with Wiley Terms and Conditions for Self-Archiving http://olabout.wiley.com/WileyCDA/Section/id-820227.html “ Enantioselective Iodine(I/III) Catalysis in Organic Synthesis Andrea Flores,a Eric Cots,a Julien Bergès,a and Kilian Muñiza,b* a Institute for Chemical Research of Catalonia (ICIQ), The Barcelona Institute of Science, Av. Països Catalans 16, 43007 Tarragona, Spain Fax: +34 977 920 224. E-mail: [email protected] b ICEA Passeig Lluís Companys, 23, 08010 Barcelona, Spain Abstract. Chiral aryliodine(III) reagents have provided an advanced concept for enantioselective synthesis and catalysis. Keywords: Chirality; Enantioselective Synthesis; With the advent of chiral iodine(I/III) catalysis, a large Homogeneous Catalysis; Iodine; Oxidation number of different structures have been explored in the area. The currently most prominent catalyst design is based on a resorcinol core and the attachment of two lactic side chains bearing ester or amide groups. It enables a privileged modular catalyst synthesis, in which fine-tuning with respect to the specific reaction requirement is straightforward. The present overview summarizes the structural variation and optimization of such chiral aryliodine catalysts and discusses structural properties of the active iodine(III) catalyst states. The status quo of enantioselective iodine(I/III) oxidation catalysis with respect to intramolecular and intermolecular reaction control is reviewed, and specific aspects of the individual catalytic cycles are discussed. -
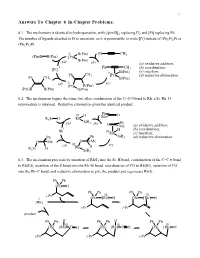
Answers to Chapter 6 In-Chapter Problems
1 Answers To Chapter 6 In-Chapter Problems. 6.1. The mechanism is identical to hydrogenation, with [(pin)B]2 replacing H2 and [Pt] replacing Pd. The number of ligands attached to Pt is uncertain, so it is permissible to write [Pt] instead of (Ph3P)2Pt or (Ph3P)3Pt. II B(Pin) Ph CH3 (Pin)B B(Pin) [Pt] B(Pin) (a) (b) (a) oxidative addition; 0 Ph CH (b) coordination; [Pt] 3 II B(Pin) (c) insertion; Ph CH3 [Pt] (d) reductive elimination. Ph CH3 B(Pin) (d) II (c) [Pt] B(Pin) (Pin)B B(Pin) B(Pin) 6.2. The mechanism begins the same, but after coordination of the C=O π bond to Rh, a Si–Rh–O intermediate is obtained. Reductive elimination gives the identical product. Ph III H Me O R3Si H Rh (a) SiR3 (b) Ph O Me (a) oxidative addition; I H (b) coordination; Rh IIIRh (c) insertion; Ph Ph SiR3 (d) reductive elimination. O Me O Me III (c) (d) Rh H R3Si H SiR3 6.3. The mechanism proceeds by insertion of Rh(I) into the Si–H bond, coordination of the C=C π bond to Rh(III), insertion of the π bond into the Rh–Si bond, coordination of CO to Rh(III), insertion of CO into the Rh–C bond, and reductive elimination to give the product and regenerate Rh(I). Ph Ph OSi H Ph Ph Ph Ph i-Pr III III I OSi [Rh] H OSi [Rh] H [Rh] i-Pr i-Pr product Ph Ph H Ph Ph H Ph Ph III III III OSi [Rh] C O OSi [Rh] C O OSi [Rh] H i-Pr i-Pr i-Pr Chapter 6 2 6.4. -

Role of Electron-Deficient Olefin Ligands in a Ni-Catalyzed Aziridine
pubs.acs.org/JACS Article Role of Electron-Deficient Olefin Ligands in a Ni-Catalyzed Aziridine Cross-Coupling To Generate Quaternary Carbons Jesuś G. Estrada, Wendy L. Williams, Stephen I. Ting, and Abigail G. Doyle* Cite This: J. Am. Chem. Soc. 2020, 142, 8928−8937 Read Online ACCESS Metrics & More Article Recommendations *sı Supporting Information ABSTRACT: We previously reported the development of an electron-deficient olefin (EDO) ligand, Fro-DO, that promotes the generation of quaternary carbon centers via Ni-catalyzed Csp3− Csp3 cross-coupling with aziridines. By contrast, electronically and structurally similar EDO ligands such as dimethyl fumarate and electron-deficient styrenes afford primarily β-hydride elimination side reactivity. Only a few catalyst systems have been identified that promote the formation of quaternary carbons via Ni-catalyzed Csp3−Csp3 cross-coupling. Although Fro-DO represents a promis- ing ligand in this regard, the basis for its superior performance is not well understood. Here we describe a detailed mechanistic study of the aziridine cross-coupling reaction and the role of EDO ligands in facilitating Csp3−Csp3 bond formation. This analysis reveals that cross-coupling proceeds by a Ni0/II cycle with a NiII azametallacyclobutane catalyst resting state. Turnover-limiting C−C reductive elimination occurs from a spectroscopically observable NiII-dialkyl intermediate bound to the EDO. Computational analysis shows that Fro-DO accelerates turnover limiting reductive elimination via LUMO lowering. However, it is no more effective than dimethyl fumarate at reducing the barrier to Csp3− Csp3 reductive elimination. Instead, Fro-DO’s unique reactivity arises from its ability to associate favorably to NiII intermediates. -

Reversible C–C Bond Formation Using Palladium Catalysis
Reversible C–C Bond Formation Using Palladium Catalysis Austin Marchese University of Toronto https://orcid.org/0000-0002-3090-2748 Bijan Mirabi University of Toronto https://orcid.org/0000-0002-4852-3439 Colton Johnson University of Toronto https://orcid.org/0000-0002-0905-1248 Mark Lautens ( [email protected] ) University of Toronto https://orcid.org/0000-0002-0179-2914 Article Keywords: C-C bonds, reactions, metal-catalyzed reactions Posted Date: May 13th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-426770/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License 1 2 Reversible C–C Bond Formation Using Palladium Catalysis 3 Authors: Austin D. Marchese1†, Bijan Mirabi1†, Colton E. Johnson1, Mark Lautens1* 4 Affiliations: 5 1Department of Chemistry, Davenport Chemical Laboratories, University of Toronto, Toronto, 6 Ontario M5S 3H6, Canada. 7 *Correspondence to: Department of Chemistry, Davenport Chemical Laboratories, University of 8 Toronto, Toronto, Ontario M5S 3H6, Canada; orcid.org/0000-0002-0179- 2914; Email: 9 [email protected]. 10 †These authors contributed equally. 11 Abstract: A widely appreciated principle is that all reactions are fundamentally reversible. 12 Observing reversible transition metal-catalyzed reactions, particularly those that include the 13 cleavage of C–C bonds, are more challenging. The development of the palladium- and nickel- 14 catalyzed carboiodination reactions afforded access to the syn- and anti-diastereomers of the 15 iodo-dihydroisoquinolone products. Using these substrates, an extensive study investigating the 16 catalytic reversibility of the C–C bond formation using a different palladium catalyst was 17 undertaken. -

Photoredox Catalysis with Metal Complexes Made from Earth-Abundant Elements
Photoredox Catalysis with Metal Complexes Made from Earth-Abundant Elements Christopher B. Larsen,[a] and Oliver S. Wenger*[a] Abstract: Photoredox chemistry with metal complexes as here, and there have been 3 prior reviews of photoredox sensitizers and catalysts frequently relies on precious elements catalysis with this particular focus.[10] However, the present such as ruthenium or iridium. Over the past 5 years, important review is thematically broader with respect to the different CuI progress towards the use of complexes made from earth- and non-CuI photoredox studies considered. It focuses on abundant elements in photoredox catalysis has been made. This organic transformations that require excitation of (usually) well review summarizes the advances made with photoactive CrIII, defined transition metal complexes that act as light-absorbers FeII, CuI, ZnII, ZrIV, Mo0 and UVI complexes in the context of and photoredox catalysts. Polymerization initiation processes, synthetic organic photoredox chemistry using visible light as an UV-light induced click reactions, and reactions relying on the energy input. Mechanistic considerations are combined with photodriven formation of catalytically active partner complexes in discussions of reaction types and scopes. Perspectives for the their electronic ground states are usually not considered. future of the field are discussed against the background of recent significant developments of new photoactive metal complexes made from earth-abundant elements. 2. Principles Photoredox behavior arises due to excited-state redox 1. Introduction properties being drastically altered from the corresponding ground state redox properties.[1] This can be understood at the Many complexes with the d6 electron configuration made from most basic level by considering for example a metal-to-ligand RuII, ReI, OsII and IrIII exhibit comparatively long-lived charge-transfer excited state (Figure 1).