Rationalization of the Mechanism of in Situ Pd(0) Formation for Cross-Coupling Reactions from Novel Unsymmetrical Pincer Palladacycles Using DFT Calculations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Oxidative Addition & Reductive Elimination
3/1/2021 Oxidative Addition & Reductive Elimination Robert H. Crabtree: Pages 159 – 182 and 235 - 266 1 Oxidative Addition Change in Example Group configuration • Oxidative addition is a key step in many transition-metal catalyzed reactions 10 8 I III • Basic reaction: d d Cu Cu 11 X X Pd0 PdII 10 LnM + LnM Pt0 PtII 10 Y Y d8 d6 PdII PdIV 10 • The new M-X and M-Y bonds are formed using the electron pair of the X-Y PtII PtIV 10 bond and one metal-centered lone pair IrI IrIII 9 RhI RhIII 9 • The metal goes up in oxidation state (+2), X-Y formally gets reduced to X-, Y- d6 d4 ReI ReIII 7 • The ease of addition (or elimination) can be tuned by the electronic and 0 II steric properties of the ancillary ligands Mo Mo 6 • OA favored by strongly e-donating L d4 d2 MoII MoIV 6 • The most common applications involve: a) Late transition metals (platinum metals: Ru, Rh, Pd, Ir, Pt) - not d7 d6 2CoII 2CoIII 9 too sensitive to O2 and H2O; routinely used in organic synthesis b) C-Halogen, H-H or Si-H bonds d4 d3 2CrII 2CrIII 6 • Common for transition metals, rare for main-group metals but: Grignard reagents! 2 1 3/1/2021 Oxidative addition – concerted mechanism Concerted addition - mostly with non-polar X-Y bonds X X X LnM + LnM LnM Y Y Y A Cr(CO)5: • H2, silanes, alkanes, ... coordinatively Cr(PMe3)5: • Arene C-H bonds more reactive than alkane C-H bonds unsaturated, but metal- centered lone pairs not phosphines are better • H-H, C-H strong bond, but M-H and M-C bonds can be very available donors, weaker acceptors full oxidative -

Relative Strength of Common Directing Groups in Palladium-Catalyzed Aromatic C-H Activation
Article Relative Strength of Common Directing Groups in Palladium-Catalyzed Aromatic CÀH Activation Anna Tomberg, Michael E´ ric Muratore, Magnus Jan Johansson, Ina Terstiege, Christian Sko¨ ld, Per-Ola Norrby per-ola.norrby@astrazeneca. com HIGHLIGHTS Directing group strength for ortho-palladation can be predicted quantum chemically Correlation with fragments allow regioselectivity predictions in complex molecules Directing strength is enhanced by deprotonation under the reaction conditions Palladation in between two directing groups is disfavored sterically; no synergy Tomberg et al., iScience 20, 373–391 October 25, 2019 ª 2019 The Author(s). https://doi.org/10.1016/ j.isci.2019.09.035 Article Relative Strength of Common Directing Groups in Palladium-Catalyzed Aromatic CÀH Activation Anna Tomberg,1 Michael E´ ric Muratore,2 Magnus Jan Johansson,2 Ina Terstiege,3 Christian Sko¨ ld,4 and Per-Ola Norrby5,6,* SUMMARY Efficient functionalization of CÀH bonds can be achieved using transition metal catalysts, such as Pd(OAc)2. To better control the regioselectivity in these reactions, some functional groups on the sub- strate may be used as directing groups, guiding the reactivity to an ortho position. Herein, we describe a methodology to score the relative strength of such directing groups in palladium-catalyzed aromatic CÀH activation. The results have been collected into a scale that serves to predict the regio- selectivity on molecules with multiple competing directing groups. We demonstrate that this scale yields accurate predictions on over a hundred examples, taken from the literature. In addition to the regioselectivity prediction on complex molecules, the knowledge of the relative strengths of directing groups can also be used to work with new combinations of functionalities, exploring un- charted chemical space. -

Roles of Base in the Pd-Catalyzed Annulative Chlorophenylene Dimerization
!"#$%&"'&()%$&*+&,-$&./01),)#23$/&4++5#),*6$& 1-#"7"8-$+2#$+$&9*:$7*3),*"+& Li-Ping Xu,a,b Brandon E. Haines,c Manjaly J. Ajitha,a Kei Murakami,d Kenichiro Itami,*,d and Djamaladdin G. Musaev*,a a Cherry L. Emerson Center for Scientific Computation, Emory University, Atlanta, Georgia 30322, USA b School of Chemistry and Chemical Engineering, Shandong University of Technology, Zibo, 255000, China c Westmont College, Department of Chemistry, Santa Barbara CA, 93108, USA d Institute of Transformative Bio-Molecules (WPI-ITbM) and Graduate School of Science, and JST-ERATO, Itami Molecular Nanocarbon Project, Nagoya University, Chikusa, Nagoya 464- 8602, Japan KEYWORDS: Palladium catalyst, polycyclic aromatic hydrocarbon, C-H functionalization, DFT, base effect ABSTRACT By using computation, the detailed mechanism of the Pd-catalyzed annulative chlorophenylene dimerization has been elucidated and the roles of the base have been identified. It is shown that the initial steps of this reaction are the active catalyst formation and the first C–Cl bond activation proceeds via the “base-assisted oxidative addition” mechanism. Overall, these steps of the reaction proceed via 16.2 kcal/mol free energy barrier for the C–Cl bond activation and are highly exergonic. Importantly, the base directly participates in the C–Cl bond cleavage introduces a mechanistic switch of the Pd(0)/Pd(II) oxidation and makes the reaction more exergonic. The following steps of the reaction are palladacycle and Pd-aryne formations, among which the palladacycle formation is found to be favorable: it proceeds with a moderate C–H activation barrier and is slightly exergonic. Although the Pd-aryne formation requires a slightly lower energy barrier, it is highly endergonic: therefore, the participation of the Pd-aryne complex, or its derivatives, in the Pd- catalyzed annulative chlorophenylene dimerization in the presence of Cs2CO3 seems to be unlikely. -

Pd-Pincer Complexes Containing Six-Membered Rings That Are More Active Allylation Catalysts Than Five-Membered Ring Analogues
Pd-pincer complexes containing six-membered rings that are more active allylation catalysts than five-membered ring analogues Dan E. Wise,a Natalie E. Pridmore,a Paul G. Pringlea aSchool of Chemistry, University of Bristol, Cantock’s Close, Bristol, BS8 1TS, U.K. Abstract A series of symmetric and non-symmetric PCP-pincer complexes have been prepared and an evaluation of their activity in catalytic allylic alkylation carried out. The X-ray crystallography and NMR spectroscopy data suggest that metallacycles containing one (or two) 6-membered rings are inherently more rigid than initially anticipated. The observed catalyst activity is found to increase as the metallacycle size increases from 5 to 6. Introduction Since the pioneering work on cyclometalated phosphine pincer complexes by Shaw et al. and amine pincer complexes by van Koten et al. in the 1970s, chemists have exploited the tunability of pincer complexes to develop many thousands of variants.1,2 The rich chemistry of pincer complexes is attributed to the many points of derivatisation present, such as the donor atoms and substituents, backbone structure, metal centre, and ancillary ligands. The applications of transition metal pincer complexes in catalysis have been extensively reviewed.3–7 Most pincer complexes are derived from C2v symmetric ligands which give rise to complexes with two fused 5-membered metallacycles (i.e. 5,5-metallacycles).3 Less well studied are pincer complexes with fused 6-membered metallacycles (i.e. 6,6-metallacycles).8–10 Pincer complexes containing 6-membered metallacycles have a larger P-M-P bite angle and would be anticipated to be more conformationally flexible. -

Palladium-Catalyzed Synthesis of Β-Hydroxy Compounds Via A
Chemical Science EDGE ARTICLE View Article Online View Journal | View Issue Palladium-catalyzed synthesis of b-hydroxy compounds via a strained 6,4-palladacycle from Cite this: Chem. Sci., 2020, 11,7260 directed C–H activation of anilines and C–O All publication charges for this article have been paid for by the Royal Society insertion of epoxides† of Chemistry a b b Raju S. Thombal, Taisiia Feoktistova, Gisela A. Gonzalez-Montiel,´ Paul H.-Y. Cheong *b and Yong Rok Lee *a A palladium-catalyzed C–H activation of acetylated anilines (acetanilides, 1,1-dimethyl-3-phenylurea, 1- phenylpyrrolidin-2-one, and 1-(indolin-1-yl)ethan-1-one) with epoxides using O-coordinating directing groups was accomplished. This C–H alkylation reaction proceeds via formation of a previously unknown 6,4-palladacycle intermediate and provides rapid access to regioselectively functionalized b-hydroxy products. Notably, this catalytic system is applicable for the gram scale mono-functionalization of acetanilide in good yields. The palladium-catalyzed coupling reaction of the ortho-C(sp2) atom of O- Creative Commons Attribution-NonCommercial 3.0 Unported Licence. Received 11th March 2020 coordinating directing groups with a C(sp3) carbon of chiral epoxides offers diverse substrate scope in Accepted 20th June 2020 good to excellent yields. In addition, further transformations of the synthesized compound led to DOI: 10.1039/d0sc01462a biologically important heterocycles. Density functional theory reveals that the 6,4-palladacycle leveraged À rsc.li/chemical-science in this work is significantly more strained (>10 kcal mol 1) than the literature known 5,4 palladacycles. -
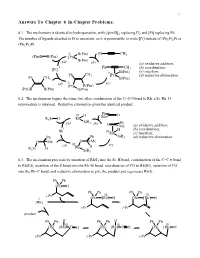
Answers to Chapter 6 In-Chapter Problems
1 Answers To Chapter 6 In-Chapter Problems. 6.1. The mechanism is identical to hydrogenation, with [(pin)B]2 replacing H2 and [Pt] replacing Pd. The number of ligands attached to Pt is uncertain, so it is permissible to write [Pt] instead of (Ph3P)2Pt or (Ph3P)3Pt. II B(Pin) Ph CH3 (Pin)B B(Pin) [Pt] B(Pin) (a) (b) (a) oxidative addition; 0 Ph CH (b) coordination; [Pt] 3 II B(Pin) (c) insertion; Ph CH3 [Pt] (d) reductive elimination. Ph CH3 B(Pin) (d) II (c) [Pt] B(Pin) (Pin)B B(Pin) B(Pin) 6.2. The mechanism begins the same, but after coordination of the C=O π bond to Rh, a Si–Rh–O intermediate is obtained. Reductive elimination gives the identical product. Ph III H Me O R3Si H Rh (a) SiR3 (b) Ph O Me (a) oxidative addition; I H (b) coordination; Rh IIIRh (c) insertion; Ph Ph SiR3 (d) reductive elimination. O Me O Me III (c) (d) Rh H R3Si H SiR3 6.3. The mechanism proceeds by insertion of Rh(I) into the Si–H bond, coordination of the C=C π bond to Rh(III), insertion of the π bond into the Rh–Si bond, coordination of CO to Rh(III), insertion of CO into the Rh–C bond, and reductive elimination to give the product and regenerate Rh(I). Ph Ph OSi H Ph Ph Ph Ph i-Pr III III I OSi [Rh] H OSi [Rh] H [Rh] i-Pr i-Pr product Ph Ph H Ph Ph H Ph Ph III III III OSi [Rh] C O OSi [Rh] C O OSi [Rh] H i-Pr i-Pr i-Pr Chapter 6 2 6.4. -

Bedford-Type Palladacycle Catalyzed Miyaura-Borylation of Aryl Halides with Tetrahydroxydiboron in Water
Bedford-type palladacycle catalyzed Miyaura-borylation of aryl halides with tetrahydroxydiboron in water Item Type Article Authors Zernickel, Anna; du, weiyuan; Ghorpade, Seema; Sawant, Dinesh; Makki, Arwa; Sekar, Nagaiyan; Eppinger, Jörg Citation Zernickel A, Du W, Ghorpade S, Sawant DN, Makki AA, et al. (2018) Bedford-type palladacycle catalyzed Miyaura-borylation of aryl halides with tetrahydroxydiboron in water. The Journal of Organic Chemistry. Available: http://dx.doi.org/10.1021/ acs.joc.7b02771. Eprint version Post-print DOI 10.1021/acs.joc.7b02771 Publisher American Chemical Society (ACS) Journal The Journal of Organic Chemistry Rights This document is the Accepted Manuscript version of a Published Work that appeared in final form in The Journal of Organic Chemistry, copyright © American Chemical Society after peer review and technical editing by the publisher. To access the final edited and published work see http://pubs.acs.org/doi/10.1021/ acs.joc.7b02771. Download date 28/09/2021 03:17:04 Link to Item http://hdl.handle.net/10754/626747 Subscriber access provided by King Abdullah University of Science and Technology Library Article Bedford-type palladacycle catalyzed Miyaura-borylation of aryl halides with tetrahydroxydiboron in water Anna Zernickel, Weiyuan Du, Seema Ghorpade, Dinesh Nanaji Sawant, Arwa A. Makki, Nagaiyan Sekar, and Jörg Eppinger J. Org. Chem., Just Accepted Manuscript • DOI: 10.1021/acs.joc.7b02771 • Publication Date (Web): 09 Jan 2018 Downloaded from http://pubs.acs.org on January 14, 2018 Just Accepted “Just Accepted” manuscripts have been peer-reviewed and accepted for publication. They are posted online prior to technical editing, formatting for publication and author proofing. -

Palladium-Catalyzed Suzuki–Miyaura Cross-Coupling in Continuous Flow
catalysts Review Palladium-Catalyzed Suzuki–Miyaura Cross-Coupling in Continuous Flow Christophe Len 1,*, Sophie Bruniaux 1, Frederic Delbecq 2 and Virinder S. Parmar 3 1 Centre de Recherche Royallieu, Université de Technologie de Compiègne (UTC), Sorbonne Universités, CS 60319, F-60203 Compiègne CEDEX, France; [email protected] 2 Ecole Supérieure de Chimie Organique et Minérale (ESCOM), 1 rue du Réseau Jean-Marie Buckmaster, F-60200 Compiègne, France; [email protected] 3 Institute of Advanced Sciences, 86-410 Faunce Corner Mall Road, Dartmouth, MA 02747, USA; [email protected] * Correspondence: [email protected]; Tel.: +33-344-234-323 Academic Editor: Ioannis D. Kostas Received: 15 March 2017; Accepted: 25 April 2017; Published: 9 May 2017 Abstract: Carbon–carbon cross-coupling reactions are among the most important processes in organic chemistry and Suzuki–Miyaura reactions are the most widely used protocols. For a decade, green chemistry and particularly catalysis and continuous flow, have shown immense potential in achieving the goals of “greener synthesis”. To date, it seems difficult to conceive the chemistry of the 21st century without the industrialization of continuous flow process in the area of pharmaceuticals, drugs, agrochemicals, polymers, etc. A large variety of palladium Suzuki–Miyaura cross-coupling reactions have been developed using a continuous flow sequence for preparing the desired biaryl derivatives. Our objective is to focus this review on the continuous flow Suzuki–Miyaura cross-coupling using homogeneous and heterogeneous catalysts. Keywords: palladium; continuous flow; Suzuki–Miyaura; cross-coupling 1. Introduction Among the main reactions in organic chemistry, C–C bond formation via a cross-coupling reaction catalyzed by transition metals is undoubtedly the most important and has been exploited very widely in the recent years. -

John F. Hartwig Henry Rapoport Professor of Chemistry
John F. Hartwig Henry Rapoport Professor of Chemistry Department of Chemistry, University of California Berkeley 718 Latimer Hall MC# 1460, Berkeley, CA 94720-1460 Email: [email protected] http://www.cchem.berkeley.edu/jfhgrp/ Personal Born August 7, 1964 in Elmhurst, IL Employment 2011-present University of California, Berkeley Henry Rapoport Professor of Chemistry. 2011-present Lawrence Berkeley National Laboratory, Berkeley Senior Faculty Scientist. 2006-2011 University of Illinois Urbana-Champaign Kenneth L. Reinhart Jr. Professor of Chemistry. 2004-2006 Yale University, New Haven, CT Irénée DuPont Professor of Chemistry. 1998-2004 Yale University, New Haven, CT Professor of Chemistry. 1996-1998 Yale University, New Haven, CT Associate Professor of Chemistry. 1992-1996 Yale University, New Haven, CT Assistant Professor of Chemistry. Appointment commenced July 1, 1992. 1990-1992 Massachusetts Institute of Technology, Cambridge, MA American Cancer Society Postdoctoral Associate. 1986-1989 University of California, Berkeley, CA Graduate Student Instructor. Taught organic chemistry to undergraduate students and inorganic chemistry to graduate students. 1985 Monsanto Japan Ltd., Kawachi, Japan Worked among an all-Japanese staff for three months on an agricultural and surface science research project. 1984 General Electric Research and Development, Schenectady, NY Synthesis of novel monomers, ionomers and polymer blends. Education 1990-1992 Massachusetts Institute of Technology, Cambridge, MA Postdoctoral Advisor: Prof. Stephen J. Lippard Studied the Pt-DNA adducts formed by an orally active platinum antitumor drug and the ability of these adducts to block DNA replication and bind cellular proteins. Designed, synthesized, and analyzed a platinum antitumor drug possessing a fluorescent ligand for in vivo monitoring. 1986-1990 University of California, Berkeley, CA Ph.D., Chemistry. -

End Functionalization of Polyisobutylenes and Their
END FUNCTIONALIZATION OF POLYISOBUTYLENES AND THEIR APPLICATIONS IN DYEING POLYOLEFINS AND IN HOMOGENEOUS CATALYSIS A Dissertation by NILUSHA PRIYADARSHANI BORALUGODAGE Submitted to the Office of Graduate Studies of Texas A&M University in Partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Chair of Committee, David E. Bergbreiter Committee Members, Daniel Singleton Karen L. Wooley Jodie L. Lutkenhaus Head of Department, David H. Russell August 2013 Major Subject: Chemistry Copyright 2013 Nilusha Priyadarshani Boralugodage ABSTRACT Non-polar phase selectively soluble polyisobutylene (PIB) is a useful polymer as a polymer support for homogeneous catalysts. Substitution of PIB onto other useful ligands alters the solubility properties of original ligands. However, such chemical modification of PIB polymers is challenging due to its limited synthetic availability. This dissertation describes the end functionalization of this terminal alkene functionalized PIB into variety of other functional groups via variety of chemistries such as electrophilic aromatic substitutions, nucleophilic aromatic substitution reactions and alkylation reactions. PIB-anilines and PIB-phenols can be prepared via electrophilic aromatic substitution reactions of anilines and phenols by PIB-alkene. Excellent oxidative and chemical resistance and nonpolar phase selective solubility of these modified PIB polymers allow these materials to be useful as soluble polymer supports for ligands and transition metal catalysts. This dissertation details immobilization of palladium and ruthenium catalysts on to these modified PIB ligands and their catalytic properties and catalyst recyclability. PIB-bound palladium catalyst was used as a recyclable homogeneous catalyst for cross coupling reactions in a thermomorphic liquid/liquid separation system- biphasic at a lower temperature and monophasic at a higher temperature. -

Role of Electron-Deficient Olefin Ligands in a Ni-Catalyzed Aziridine
pubs.acs.org/JACS Article Role of Electron-Deficient Olefin Ligands in a Ni-Catalyzed Aziridine Cross-Coupling To Generate Quaternary Carbons Jesuś G. Estrada, Wendy L. Williams, Stephen I. Ting, and Abigail G. Doyle* Cite This: J. Am. Chem. Soc. 2020, 142, 8928−8937 Read Online ACCESS Metrics & More Article Recommendations *sı Supporting Information ABSTRACT: We previously reported the development of an electron-deficient olefin (EDO) ligand, Fro-DO, that promotes the generation of quaternary carbon centers via Ni-catalyzed Csp3− Csp3 cross-coupling with aziridines. By contrast, electronically and structurally similar EDO ligands such as dimethyl fumarate and electron-deficient styrenes afford primarily β-hydride elimination side reactivity. Only a few catalyst systems have been identified that promote the formation of quaternary carbons via Ni-catalyzed Csp3−Csp3 cross-coupling. Although Fro-DO represents a promis- ing ligand in this regard, the basis for its superior performance is not well understood. Here we describe a detailed mechanistic study of the aziridine cross-coupling reaction and the role of EDO ligands in facilitating Csp3−Csp3 bond formation. This analysis reveals that cross-coupling proceeds by a Ni0/II cycle with a NiII azametallacyclobutane catalyst resting state. Turnover-limiting C−C reductive elimination occurs from a spectroscopically observable NiII-dialkyl intermediate bound to the EDO. Computational analysis shows that Fro-DO accelerates turnover limiting reductive elimination via LUMO lowering. However, it is no more effective than dimethyl fumarate at reducing the barrier to Csp3− Csp3 reductive elimination. Instead, Fro-DO’s unique reactivity arises from its ability to associate favorably to NiII intermediates. -

Palladium-Catalyzed Functionalization of C-H Bonds and Alkenes
Palladium-Catalyzed Functionalization of C-H Bonds and Alkenes by Lopa Vrushank Desai A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Chemistry) in The University of Michigan 2008 Doctoral Committee: Associate Professor Melanie S. Sanford, Chair Professor Edwin Vedejs Professor Erdogan Gulari Assistant Professor John P. Wolfe © Lopa Vrushank Desai ______________________ 2008 To Dr. Sanford, Dipa and Smitesh ii Acknowledgements I give my deepest gratitude to all those people who made my graduate experience a memorable and one that I will cherish forever. I would like to start with the most influential person – Dr. Sanford. I consider myself very fortunate that I got an advisor like you. Your passion, enthusiasm, motivation, dedication, patience and mentorship were paramount in my growth and life. You showed me different ways to approach a problem and guided me when I made mistakes. You taught me how to question, write and present chemistry. Even though you had a lot on your plate; you still devoted endless time to me. You are an amazing mentor because you make your students your priority – you care for us, take time to understand us, go out of your way to build our future and love us for who we are. I thank you for everything you have done for me. I hope that you will continue to be a significant part of my life. I would like to thank my committee members for their support and feedback in such short notice. Dr. Wolfe, I have learned a lot from you through group meetings and classes.