Genetic Diversity and Population Structure: Implications for Conservation of Wild Soybean (Glycine Soja Sieb
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Report on Domestic Animal Genetic Resources in China
Country Report for the Preparation of the First Report on the State of the World’s Animal Genetic Resources Report on Domestic Animal Genetic Resources in China June 2003 Beijing CONTENTS Executive Summary Biological diversity is the basis for the existence and development of human society and has aroused the increasing great attention of international society. In June 1992, more than 150 countries including China had jointly signed the "Pact of Biological Diversity". Domestic animal genetic resources are an important component of biological diversity, precious resources formed through long-term evolution, and also the closest and most direct part of relation with human beings. Therefore, in order to realize a sustainable, stable and high-efficient animal production, it is of great significance to meet even higher demand for animal and poultry product varieties and quality by human society, strengthen conservation, and effective, rational and sustainable utilization of animal and poultry genetic resources. The "Report on Domestic Animal Genetic Resources in China" (hereinafter referred to as the "Report") was compiled in accordance with the requirements of the "World Status of Animal Genetic Resource " compiled by the FAO. The Ministry of Agriculture" (MOA) has attached great importance to the compilation of the Report, organized nearly 20 experts from administrative, technical extension, research institutes and universities to participate in the compilation team. In 1999, the first meeting of the compilation staff members had been held in the National Animal Husbandry and Veterinary Service, discussed on the compilation outline and division of labor in the Report compilation, and smoothly fulfilled the tasks to each of the compilers. -

Effects of Cutting Frequency on Alfalfa Yield and Yield Components in Songnen Plain, Northeast China
African Journal of Biotechnology Vol. 11(21), pp. 4782-4790, 13 March, 2012 Available online at http://www.academicjournals.org/AJB DOI: 10.5897/AJB12.092 ISSN 1684–5315 © 2012 Academic Journals Full Length Research Paper Effects of cutting frequency on alfalfa yield and yield components in Songnen Plain, Northeast China Ji-shan Chen, Fen-lan Tang, Rui-fen Zhu ,Chao Gao, Gui-li Di and Yue-xue Zhang* Institute of Pratacultural Science, Heilongjiang Academy of Agricultural Sciences, Harbin, Heilongjiang 150086, China. Accepted 27 February, 2012 The productivity and quality of alfalfa ( Medicago sativa L.) is strongly influenced by cutting frequency (F). To clarify that the yield and quality of alfalfa if affected by F, an experiment was conducted on Songnen Plain in Northeast China to investigate the responses of yield components and quality to 3 cutting frequencies (F30, F40 and F60) among 3 cultivars (C) (Longmu, Aohan, Zhaodong). Result from two consecutive years showed that cutting frequency had a greater effect on reducing forage yield and yield components at F40. Cultivars had no effect on 2-year total forage yield and alfalfa quality. Alfalfa at the F40 always had higher crude protein (CP), and neutral detergent fibre (NDF) than F30 and F60 treatments. The interaction (C×F) on forage yield and components (CP and NDF) was not significant. This study provides evidence that: i) 40-day intervals can be advocated for cultivars growing in North- east China; and ii) at F40 utilization, Longmu is a well-adapted alfalfa variety in the Songnen Plain because of higher yield and quality under three cuttings. -
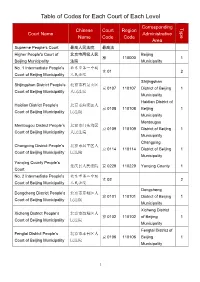
Table of Codes for Each Court of Each Level
Table of Codes for Each Court of Each Level Corresponding Type Chinese Court Region Court Name Administrative Name Code Code Area Supreme People’s Court 最高人民法院 最高法 Higher People's Court of 北京市高级人民 Beijing 京 110000 1 Beijing Municipality 法院 Municipality No. 1 Intermediate People's 北京市第一中级 京 01 2 Court of Beijing Municipality 人民法院 Shijingshan Shijingshan District People’s 北京市石景山区 京 0107 110107 District of Beijing 1 Court of Beijing Municipality 人民法院 Municipality Haidian District of Haidian District People’s 北京市海淀区人 京 0108 110108 Beijing 1 Court of Beijing Municipality 民法院 Municipality Mentougou Mentougou District People’s 北京市门头沟区 京 0109 110109 District of Beijing 1 Court of Beijing Municipality 人民法院 Municipality Changping Changping District People’s 北京市昌平区人 京 0114 110114 District of Beijing 1 Court of Beijing Municipality 民法院 Municipality Yanqing County People’s 延庆县人民法院 京 0229 110229 Yanqing County 1 Court No. 2 Intermediate People's 北京市第二中级 京 02 2 Court of Beijing Municipality 人民法院 Dongcheng Dongcheng District People’s 北京市东城区人 京 0101 110101 District of Beijing 1 Court of Beijing Municipality 民法院 Municipality Xicheng District Xicheng District People’s 北京市西城区人 京 0102 110102 of Beijing 1 Court of Beijing Municipality 民法院 Municipality Fengtai District of Fengtai District People’s 北京市丰台区人 京 0106 110106 Beijing 1 Court of Beijing Municipality 民法院 Municipality 1 Fangshan District Fangshan District People’s 北京市房山区人 京 0111 110111 of Beijing 1 Court of Beijing Municipality 民法院 Municipality Daxing District of Daxing District People’s 北京市大兴区人 京 0115 -

Laogai Handbook 劳改手册 2007-2008
L A O G A I HANDBOOK 劳 改 手 册 2007 – 2008 The Laogai Research Foundation Washington, DC 2008 The Laogai Research Foundation, founded in 1992, is a non-profit, tax-exempt organization [501 (c) (3)] incorporated in the District of Columbia, USA. The Foundation’s purpose is to gather information on the Chinese Laogai - the most extensive system of forced labor camps in the world today – and disseminate this information to journalists, human rights activists, government officials and the general public. Directors: Harry Wu, Jeffrey Fiedler, Tienchi Martin-Liao LRF Board: Harry Wu, Jeffrey Fiedler, Tienchi Martin-Liao, Lodi Gyari Laogai Handbook 劳改手册 2007-2008 Copyright © The Laogai Research Foundation (LRF) All Rights Reserved. The Laogai Research Foundation 1109 M St. NW Washington, DC 20005 Tel: (202) 408-8300 / 8301 Fax: (202) 408-8302 E-mail: [email protected] Website: www.laogai.org ISBN 978-1-931550-25-3 Published by The Laogai Research Foundation, October 2008 Printed in Hong Kong US $35.00 Our Statement We have no right to forget those deprived of freedom and 我们没有权利忘却劳改营中失去自由及生命的人。 life in the Laogai. 我们在寻求真理, 希望这类残暴及非人道的行为早日 We are seeking the truth, with the hope that such horrible 消除并且永不再现。 and inhumane practices will soon cease to exist and will never recur. 在中国,民主与劳改不可能并存。 In China, democracy and the Laogai are incompatible. THE LAOGAI RESEARCH FOUNDATION Table of Contents Code Page Code Page Preface 前言 ...............................................................…1 23 Shandong Province 山东省.............................................. 377 Introduction 概述 .........................................................…4 24 Shanghai Municipality 上海市 .......................................... 407 Laogai Terms and Abbreviations 25 Shanxi Province 山西省 ................................................... 423 劳改单位及缩写............................................................28 26 Sichuan Province 四川省 ................................................ -

Journalist Biographie Archibald, John
Report Title - p. 1 of 303 Report Title Amadé, Emilio Sarzi (Curtatone 1925-1989 Mailand) : Journalist Biographie 1957-1961 Emilio Sarzi Amadé ist Korrespondent für Italien in China. [Wik] Archibald, John (Huntley, Aberdeenshire 1853-nach 1922) : Protestantischer Missionar, Journalist Biographie 1876-1913 John Archibald arbeitet für die National Bible Society of Scotland in Hankou. Er resit in Hubei, Hunan, Henan, Anhui und Jiangxi. [Who2] 1913 John Archibald wird Herausgeber der Central China post. [Who2] Bibliographie : Autor 1910 Archibald, John. The National Bible Society of Scotland. In : The China mission year book ; Shanghai (1910). [Int] Balf, Todd (um 2000) : Amerikanischer Journalist, Senior Editor Outside Magazine, Mitherausgeber Men's journal Bibliographie : Autor 2000 Balf, Todd. The last river : the tragic race for Shangi-la. (New York, N.Y. : Crown, 2000). [Erstbefahrung 1998 für die National Geographic Society durch wilde Schluchten des Brahmaputra (Tsangpo) in Tibet, die wegen Strömungen und Tod von Douglas Gordon (1956-1998) scheitert]. [WC,Cla] Balfour, Frederic Henry (1846-1909) : Kaufmann, Journalist in China Bibliographie : Autor 1876 Balfour, Frederic Henry. Waifs and strays from the Far East ; being a series of disconnected essays on matters relating to China. (London : Trübner, 1876). https://archive.org/details/waifsstraysfromf00balfrich. 1881 Chuang Tsze. The divine classsic of Nan-hua : being the works of Chuang Tsze, taoist philosopher. With an excursus, and copious annotations in English and Chinese by Frederic Henry Balfour. (Shanhgai ; Hongkong : Kelly & Walsh, 1881). [Zhuangzi. Nan hua jing]. https://catalog.hathitrust.org/Record/100328385. 1883 Balfour, Frederic Henry. Idiomatic dialogues in the Peking colloquial for the use of students. (Shanghai : Printed at the North-China Herald Office, 1883). -

The Analysis of Human Factors on Grassland Productivity in Western Songnen Plain
View metadata, citation and similar papers at core.ac.uk brought to youCORE by provided by Elsevier - Publisher Connector Available online at www.sciencedirect.com Procedia Environmental Sciences 10 ( 2011 ) 1302 – 1307 2011 3rd International Conference on Environmental Science and InformationConference Application Title Technology (ESIAT 2011) The Analysis of Human Factors on Grassland Productivity in Western Songnen Plain Shufeng Zheng, Yuan Sun* College of Agricultural Resources and Environment, Heilongjiang University, P.O. Box 184, 74 Xuefu Road, Nangang Distrit, Harbin, 150080, P. R. China Abstract West Songnen Plain is ecologically fragile area and degrading ecosystem. Over the past 50 years, interfered by the natural factors and human activities, the quality of grassland and the carrying capacity declined. It is important for the rational utilization of grassland resources and the carbon cycle study of terrestrial ecosystem by analyzing human factors on the grassland productivity. The studying sites were divided into 8 land units with relatively homogeneous natural conditions. Then identify the grassland areas interfered and those not interfered by human activities. The sum NDVI of each land unit were obtained by using satellite remote sensing data. The effects of human factors on grassland productivity were got through calculating the relative degradation index of grassland. It showed that the productivity of grassland without anthropogenic interference was much higher than that of anthropogenic interference in the growing season. The impact of human factors on grassland was smaller in west of Songnen Plain in 2002 and 2005, but bigger in 2004. There is no obvious correlativity between the climatic factors and grassland relative degradation index in west of Songnen Plain. -

China Linen Textile Industry, Ltd. (OTC BB: CTXIF)
China Linen Textile Industry, Ltd. (OTC BB: CTXIF) TMTM APRIL 12, 2010 | TARGET PRICE: $4.00 | RATING: BUY DISCOVERING TOMORROW’S BLUE CHIPS TODAY TM Visibility COMPANY OVERVIEW China Linen Textile Industry Ltd. (“CTXIF” or the “Company”) is a leading I NITIAL REPORT China-based manufacturer and distributor of linen products. The Company manufactures over 50 types of linen yarn and 100 varieties of linen fabric and ANALYST products. Roughly 55% of CTXIF’s production is exported to Europe, North Aditya Khandekar, CFA America, the Middle East and South America with the remainder sold in China. The Company enjoys a 5% market share of China’s total linen industry and MARKET DATA ranks among the top 5 companies in China’s linen textile industry. TICKER CTXIF FISCAL YEAR DECEMBER INVESTMENT RATIONALE LINEN/ SECTOR TEXTILES Strong near-term revenue growth and operating performance projected. RECENT PRICE $1.67 CTXIF has posted annual revenue and net profi t growth of about 40% on TARGET PRICE $4.00 average for the last fi ve years. The Company has maintained a healthy EBITDA MARKET CAP $33.7M margin of greater than 19% in the last few years with return on assets greater 52-WEEK HIGH - LOW $1.99 - $0.15 than 10%. The Company is running its plants at peak utilization and still PRICE/EARNINGS (TTM) 5.6X requires outsourced production to meet excess demand. With capacity additions PRICE/BOOK (MRQ) 1.6X expected in the next few years, we are projecting 20% year-over-year growth in PRICE/SALES (TTM) 1.1X the short-term and 15% year-over-year growth in the mid-term. -

中國內地指定醫院列表 出版日期: 2019 年 7 月 1 日 Designated Hospital List in Mainland China Published Date: 1 Jul 2019
中國內地指定醫院列表 出版日期: 2019 年 7 月 1 日 Designated Hospital List in Mainland China Published Date: 1 Jul 2019 省 / 自治區 / 直轄市 醫院 地址 電話號碼 Provinces / 城市/City Autonomous Hospital Address Tel. No. Regions / Municipalities 中國人民解放軍第二炮兵總醫院 (第 262 醫院) 北京 北京 西城區新街口外大街 16 號 The Second Artillery General Hospital of Chinese 10-66343055 Beijing Beijing 16 Xinjiekou Outer Street, Xicheng District People’s Liberation Army 中國人民解放軍總醫院 (第 301 醫院) 北京 北京 海澱區復興路 28 號 The General Hospital of Chinese People's Liberation 10-82266699 Beijing Beijing 28 Fuxing Road, Haidian District Army 北京 北京 中國人民解放軍第 302 醫院 豐台區西四環中路 100 號 10-66933129 Beijing Beijing 302 Military Hospital of China 100 West No.4 Ring Road Middle, Fengtai District 中國人民解放軍總醫院第一附屬醫院 (中國人民解 北京 北京 海定區阜成路 51 號 放軍 304 醫院) 10-66867304 Beijing Beijing 51 Fucheng Road, Haidian District PLA No.304 Hospital 北京 北京 中國人民解放軍第 305 醫院 西城區文津街甲 13 號 10-66004120 Beijing Beijing PLA No.305 Hospital 13 Wenjin Street, Xicheng District 北京 北京 中國人民解放軍第 306 醫院 朝陽區安翔北里 9 號 10-66356729 Beijing Beijing The 306th Hospital of PLA 9 Anxiang North Road, Chaoyang District 中國人民解放軍第 307 醫院 北京 北京 豐台區東大街 8 號 The 307th Hospital of Chinese People’s Liberation 10-66947114 Beijing Beijing 8 East Street, Fengtai District Army 中國人民解放軍第 309 醫院 北京 北京 海澱區黑山扈路甲 17 號 The 309th Hospital of Chinese People’s Liberation 10-66775961 Beijing Beijing 17 Heishanhu Road, Haidian District Army 中國人民解放軍第 466 醫院 (空軍航空醫學研究所 北京 北京 海澱區北窪路北口 附屬醫院) 10-81988888 Beijing Beijing Beiwa Road North, Haidian District PLA No.466 Hospital 北京 北京 中國人民解放軍海軍總醫院 (海軍總醫院) 海澱區阜成路 6 號 10-66958114 Beijing Beijing PLA Naval General Hospital 6 Fucheng Road, Haidian District 北京 北京 中國人民解放軍空軍總醫院 (空軍總醫院) 海澱區阜成路 30 號 10-68410099 Beijing Beijing Air Force General Hospital, PLA 30 Fucheng Road, Haidian District 中華人民共和國北京市昌平區生命園路 1 號 北京 北京 北京大學國際醫院 Yard No.1, Life Science Park, Changping District, Beijing, 10-69006666 Beijing Beijing Peking University International Hospital China, 東城區南門倉 5 號(西院) 5 Nanmencang, Dongcheng District (West Campus) 北京 北京 北京軍區總醫院 10-66721629 Beijing Beijing PLA. -

Copy / Paste the Company's Name of This List Into the Relevant Datafield of Our Webpage by Using the Before Mentioned Link
List of Operators subject to the organic control system according to Commission Regulations (EC) No 1235/2008 Article 11 (3e) and equivalent to (EC) No 834/2007, (EC) No 889/2008. This list has been updated bx Kiwa BCS on 22.04.2021 This list targets at providing information without any legally commitment. Only the Operators' current Certificate is legally binding. For any further questions related to the certification status of any EU-organic Operator certified by Kiwa BCS please contact https://www.kiwa.com/de/de/aktuelle-angelegenheiten/zertifikatssuche/ [email protected] copy / paste the Company's name of this list into the relevant datafield of our webpage by using the before mentioned link. Company Name Location Country Products Status 4 Elementos Industria Barueri BRAZIL Acai, Frozen Foods Certified Alimentos 854 Community Shunli Oil 158403 Hulin City, Heilongjiang CHINA Soybean meal Certified Processing Plant Province Absolute Organix Birnham Park, Gauteng ZA Suedafrika Products as per attachment Certified AÇAÍ AMAZONAS INDUSTRIA OBIDOS, PARA BRAZIL Acai coarse 14% (or special) 84 t; Acai Fine 8% (or Popular) 84 t; Certified E COMERCIO LTDA. Acai powder 1 t; Acai powder 100% pure RWD 1 t; Acerola powder 1 t; acerola powder RWD 1 t; Camu Camu Powder 2 t; Camu Camu Powder RWD 1 t; Camu Camu pulp 0,7 t; Graviola powder 1 t ; Graviola Powdered RWD 1 t; medium acai 11% - 84 t; medium acai 12% - 84 t; Passion fruit powder RWD 1 t; Passion fruit powder 1 t; powder Mango 1 t; powdered cupuaçu 1 t; Powdered cupuaçu RWD 1 t; powdered Mango RWD 1 t; Premix 80/20 Açaí Powder 2 t; Strawberry powder 1 t; Strawberry powder RWD 1 t ADPP Bissorá, Oio GW Guinea-Bissau Cashew nuts, Cashew nuts, raw with shell Certified AGA Armazéns Gerais Araxá Araxá BRAZIL Coffee Beans, Green (3000t) Certified Ltda. -

2011 Annual Report 2011
中國航空科技工業股份有限公司 年 報 ANNUAL REPORT 2011 2011 ANNUAL REPORT 2011 中國航空科技工業股份有限公司 年報 中國航空科技工業股份有限公司 (在中華人民共和國註冊成立的股份有限公司) (A joint stock limited company incorporated in the People’s Republic of China with limited liability) ( 股 票 編 號 : 2357) (Stock Code: 2357) Designed & Produced by HeterMedia Services Limited 設計及製作由軒達資訊服務有限公司提供 AviChina Industry & Technology Company Limited Contents 2 Company Profile 4 Financial Highlights 7 Chairman’s Statement 9 Management Discussion and Analysis 22 Directors, Supervisors and Senior Management 35 Reportf o the Board 49 Reportf o the Supervisory Committee 50 Corporate Governance Report 59 Independent Auditor’s Report 61 Consolidated Income Statement 62 Consolidated Statement of Comprehensive Income 63 Balance Sheets 65 Consolidated Statement of Changes in Equity 67 Consolidated Cash Flow Statement 69 Noteso t the Financial Statements 143 Definitions 147 Corporate Information 2011 Annual Report 1 AviChina Industry & Technology Company Limited Company Profile AviChina Industry & Technology Company Limited (the “Company”) is a joint stock limited company established in the People’s Republic of China (“PRC”) on 30 April 2003. The Company’s H Shares have been listed on the Stock Exchange since 30 October 2003. The stock code of the Company is “2357”. The principal shareholders of the Company’s domestic shares are AVIC, AMES, China Hua Rong Asset Management Corporation, China Cinda Asset Management Corporation and China Orient Asset Management Corporation, and the substantial shareholder of the Company’s H shares is European Aeronautics Defence and Space Company (the “EADS”). The Company principally operates through its subsidiaries. The Company and its subsidiaries (the “Group”) are mainly engaged in: • the development, manufacture, sales and upgrade of aviation products such as helicopters, trainers, general-purpose aircraft and regional jets for domestic and overseas customers; and • the co-development and manufacture of aviation products with foreign aviation products manufacturers. -

Rapid Extraction of Regional-Scale Agricultural Disasters by the Standardized Monitoring Model Based on Google Earth Engine
sustainability Article Rapid Extraction of Regional-scale Agricultural Disasters by the Standardized Monitoring Model Based on Google Earth Engine Zhengrong Liu 1, Huanjun Liu 1,2, Chong Luo 2, Haoxuan Yang 3 , Xiangtian Meng 1, Yongchol Ju 4 and Dong Guo 2,* 1 School of Pubilc Adminstration and Law, Northeast Agricultural University, Harbin 150030, China; [email protected] (Z.L.); [email protected] (H.L.); [email protected] (X.M.) 2 Northeast Institute of Geography and Agroecology Chinese Academy of Sciences, Changchun 130102, China; [email protected] 3 College of Surveying and Geo-Informatics, Tongji University, 1239 Siping Road, Shanghai 200092, China; [email protected] 4 Wonsan University of Agriculture, Won San City, Kangwon Province, DPRK; [email protected] * Correspondence: [email protected]; Tel.: +86-1874-519-4393 Received: 15 July 2020; Accepted: 6 August 2020; Published: 12 August 2020 Abstract: Remote sensing has been used as an important tool for disaster monitoring and disaster scope extraction, especially for the analysis of spatial and temporal disaster patterns of large-scale and long-duration series. Google Earth Engine provides the possibility of quickly extracting the disaster range over a large area. Based on the Google Earth Engine cloud platform, this study used MODIS vegetation index products with 250-m spatial resolution synthesized over 16 days from the period 2005–2019 to develop a rapid and effective method for monitoring disasters across a wide spatiotemporal range. Three types of disaster monitoring and scope extraction models are proposed: the normalized difference vegetation index (NDVI) median time standardization model (RNDVI_TM(i)), the NDVI median phenology standardization model (RNDVI_AM(i)(j)), and the NDVI median spatiotemporal standardization model (RNDVI_ZM(i)(j)). -

附录I 黑龙江省目前统计到的外来入侵植物名录appendix I the Checklist Of
附录I 黑龙江省目前统计到的外来入侵植物名录 Appendix I The checklist of invasive plants recorded in Heilongjiang Province 科名 种名 原产地 传入途径 用途 生活型 分布地点 凭证标本 Family Species Origin Introduction pathway Function Life form Distribution Specimen 桑科 大麻 Cannabis sativa 亚洲中部 Central Asia II 经济 一年生草本 哈尔滨市、兰西县 陶雷134 Moraceae Economic Annual herb Harbin City and Lanxi County LeiTao134 石竹科 麦仙翁 Agrostemma githago 欧洲 Europe UI 杂草 一年生草本 牡丹江市、佳木斯市 资料记载 Caryophyllaceae Weeds Annual herb Mudanjiang and Jiamusi Data cities 肥皂草 Saponaria officinalis 欧洲 Europe UI 经济 多年生草本 哈尔滨市 Harbin City 郑宝江378 Economic Perennial herb Baojiang Zheng378 狗筋麦瓶草 Silene vulgaris 欧洲 Europe UI 杂草 多年生草本 大兴安岭地区 郑宝江734 Weeds Perennial herb Daxinganling Baojiang Zheng734 藜科 杂配藜 Chenopodium hybridum 欧洲及西亚 UI 杂草 一年生草本 全省分布 陶雷792 Chenopodiaceae Europe and western Asia Weeds Annual herb Heilongjiang Province LeiTao792 苋科 凹头苋 Amaranthus blitum 美洲热带 Tropical America UI 杂草 一年生草本 全省分布 郑宝江641 Amaranthaceae Weeds Annual herb Heilongjiang Province Baojiang Zheng641 反枝苋 A. retroflexus 美洲热带 Tropical America II 饲草 一年生草本 全省分布 郑宝江639 Forage grass Annual herb Heilongjiang Province Baojiang Zheng639 苋 A. tricolor 热带亚洲 Tropical Asia II 蔬菜 一年生草本 全省分布 郑宝江1148 Vegetables Annual herb Heilongjiang Province Baojiang Zheng1148 十字花科 密花独行菜 Lepidium densiflorum 北美洲 North America UI 杂草 一年生草本 全省分布 陶林1023 Cruciferae Weeds Annual herb Heilongjiang Province LinTao1023 豆科 紫苜蓿 Medicago sativa 西亚 West Aisa II 饲草 一年生草本 肇东市 陶林1045 Leguminosae Forage grass Annual herb Zhaodong City LinTao1045 白花草木樨 Melilotus albus 欧洲及亚洲西部 II 饲草 一年生草本 大庆市 陶林1376 Europe and western Aisa Forage grass Annual herb Daqing City LinTao1376 红车轴草 Trifolium pratense 欧洲 Europe II 饲草 多年生草本 尚志市 郑宝江648 Forage grass Perennial herb Shangzhi City Baojiang Zheng648 白车轴草 T.