Antagonism of Lidocaine Inhibition by Open-Channel Blockers That Generate Resurgent Na Current
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Topical Therapy As a Treatmentfor Brachioradial
Journal of Case Reports: Open Access Case report Open Access Topical Therapy as a Treatmentfor Brachioradial Pruritis: a Case Report Brianna De Souza M.D, Amy McMichael M.D* Department of Dermatology, Wake Forest University School of Medicine, Winston-Salem, North Carolina *Corresponding author: Amy McMichael, MD Department of Dermatology, Wake Forest Baptist Medical Center,1 Medical Center Blvd, Winston-Salem, NC 27157, Phone: 336-716-7882, Email: [email protected] Received Date: April 12, 2019 Accepted Date: May 06, 2019 Published Date: May 08, 2019 Citation: Brianna De Souza (2019) Topical Therapy as a Treatmentfor Brachioradial Pruritis: a Case Report. Case Reports: Open Access 4: 1-5. Abstract Management of brachioradial pruritus (BRP) presents a formidable challenge to dermatologists and neurologists. BRP is a rare, neurocutaneous condition characterized by sharply localized, chronic pain with associated itching, burning, stinging, and or tingling sensation. Effective care of this patient population is confounded by limitations within the litera- ture, comprised of case series and case reports. We present a case of one middle-aged female with a chronic history of BRP recalcitrant to the following oral therapies: pregabalin, gabapentin, mirtazapine, prednisone, and amitriptyline, as well as topical triamcinolone. After being evaluated in the clinic, the patient was started on combination therapy withKetamine 10%, Amitriptyline 5%, and Lidocaine 5% topical cream to which she responded. Keywords: Brachioradial pruritus, Brachioradial, Pruritus, Neurocutaneous ©2019 The Authors. Published by the JScholar under the terms of the Crea- tive Commons Attribution License http://creativecommons.org/licenses/ by/3.0/, which permits unrestricted use, provided the original author and source are credited. -
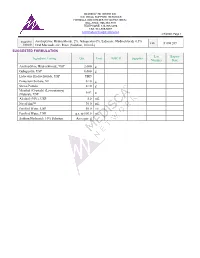
Amitriptyline Hydrochloride 2%, Gabapentin 6%, Lidocaine Hydrochloride 0.5% FIN F 008 269 Formula Oral Mucoadhesive Rinse (Solution, 100 Ml)
MEDISCA® NETWORK INC. TECHNICAL SUPPORT SERVICES FORMULATION CHEMISTRY DEPARTMENT TOLL-FREE: 866-333-7811 TELEPHONE: 514-905-5096 FAX: 514-905-5097 [email protected] 4/7/2020; Page 1 Suggested Amitriptyline Hydrochloride 2%, Gabapentin 6%, Lidocaine Hydrochloride 0.5% FIN F 008 269 Formula Oral Mucoadhesive Rinse (Solution, 100 mL) SUGGESTED FORMULATION Lot Expiry Ingredient Listing Qty. Unit NDC # Supplier Number Date Amitriptyline Hydrochloride, USP 2.000 g Gabapentin, USP 6.000 g Lidocaine Hydrochloride, USP TBD Potassium Sorbate, NF 0.10 g Stevia Powder 0.10 g Menthol (Crystals) (Levorotatory) 0.02 g (Natural), USP Alcohol (95%), USP 5.0 mL NovaFilm™ 30.0 mL Purified Water, USP 50.0 mL Purified Water, USP q.s. to 100.0 mL Sodium Hydroxide 10% Solution As required MEDISCA® NETWORK INC. TECHNICAL SUPPORT SERVICES FORMULATION CHEMISTRY DEPARTMENT TOLL-FREE: 866-333-7811 TELEPHONE: 514-905-5096 FAX: 514-905-5097 [email protected] 4/7/2020; Page 2 Suggested Amitriptyline Hydrochloride 2%, Gabapentin 6%, Lidocaine Hydrochloride 0.5% FIN F 008 269 Formula Oral Mucoadhesive Rinse (Solution, 100 mL) SPECIAL PREPARATORY CONSIDERATIONS Ingredient-Specific Information Light Sensitive (protect from light whenever possible): Amitriptyline Hydrochloride, Gabapentin Hygroscopic (protect from moisture whenever possible): Stevia Powder Narrow Therapeutic Index Lidocaine Hydrochloride Suggested Preparatory Guidelines ■ Non-Sterile Preparation □ Sterile Preparation Processing Error / To account for processing error and pH testing considerations during preparation, it is Testing Considerations: suggested to measure an additional 3 to 5% of the required quantities of ingredients. Special Instruction: This formula may contain one or more Active Pharmaceutical Ingredients (APIs) that may be classified as hazardous, please refer & verify the current NIOSH list of Antineoplastic and Other Hazardous Drugs in Healthcare Settings, 2016. -

The Effects of Lidocaine and Mefenamic Acid on Post-Episiotomy
Shiraz E-Med J. 2016 March; 17(3):e36286. doi: 10.17795/semj36286. Published online 2016 March 27. Research Article The Effects of Lidocaine and Mefenamic Acid on Post-Episiotomy Pain: A Comparative Study Masoumeh Delaram,1,* Lobat Jafar Zadeh,2 and Sahand Shams3 1Faculty of Nursing and Midwifery, Shahrekord University of Medical Sciences, Shahrekord, IR Iran 2Faculty of Medicine, Shahrekord University of Medical Sciences, Shahrekord, IR Iran 3Faculty of Veterinary Medicine, Shahrekord University, Shahrekord, IR Iran *Corresponding author: Masoumeh Delaram, Faculty of Nursing and Midwifery, Shahrekord University of Medical Sciences, Shahrekord, IR Iran. Tel: +98-3813335648, Fax: +98-3813346714, E-mail: [email protected] Received 2016 January 13; Revised 2016 February 29; Accepted 2016 March 04. Abstract Background: Most women suffer pain following an episiotomy and oral non-steroidal anti-inflammatory drugs are commonly used for pain relief. Due to the gastrointestinal side effects of oral drugs, it seems that women are more accepting of topical medications for pain relief. Objectives: Therefore, the aim of this study was to compare the effects of lidocaine and mefenamic acid on post-episiotomy pain. Patients and Methods: This clinical trial was carried out in 2011. It involved sixty women with singleton pregnancy who were given an episiotomy at 38 to 42 weeks of gestation. The participants were randomly divided into two groups. One group received 2% lido- caine cream (n = 30), while the other group received 250 mg of mefenamic acid (n = 30). The data were collected via a questionnaire and a visual analog scale. Pain intensity was compared from the first complaint by the mother and at 6, 12, and 24 hours after the delivery in both groups. -

Fenestrations Control Resting-State Block of a Voltage-Gated Sodium
Fenestrations control resting-state block of a voltage- gated sodium channel Tamer M. Gamal El-Dina,1, Michael J. Lenaeusa,b,1, Ning Zhenga,c,2,3, and William A. Catteralla,2,3 aDepartment of Pharmacology, University of Washington, Seattle, WA 98195; bDivision of General Internal Medicine, Department of Medicine, University of Washington, Seattle, WA 98195; and cHoward Hughes Medical Institute, University of Washington, Seattle, WA 98195 Contributed by William A. Catterall, October 31, 2018 (sent for review September 4, 2018; reviewed by Ryan Hibbs, Michael C. Sanguinetti, and Joerg Striessnig) Potency of drug action is usually determined by binding to a specific Receptor Hypothesis posits that resting-state block is mediated receptor site on target proteins. In contrast to this conventional by drug entry from the lipid phase of the membrane into the drug paradigm, we show here that potency of local anesthetics (LAs) and receptor site in the pore, and rapid open-state block occurs as the antiarrhythmic drugs (AADs) that block sodium channels is con- drug enters the open pore from the cytoplasm (15). Both of these trolled by fenestrations that allow drug access to the receptor site forms of block are enhanced when the channel enters the inac- directly from the membrane phase. Voltage-gated sodium channels tivated state, which has high affinity for bound drug (15). It is initiate action potentials in nerve and cardiac muscle, where their well-established that LAs and AADs can reach their binding site hyperactivity causes pain and cardiac arrhythmia, respectively. LAs from the intracellular side if the activation gate is open, which and AADs selectively block sodium channels in rapidly firing nerve gives open-state block, and that inactivation increases the affinity and muscle cells to relieve these conditions. -

Anaesthetic Implications of Calcium Channel Blockers
436 Anaesthetic implications of calcium channel Leonard C. Jenkins aA MD CM FRCPC blockers Peter J. Scoates a sc MD FRCPC CONTENTS The object of this review is to emphasize the anaesthetic implications of calcium channel block- Physiology - calcium/calcium channel blockers Uses of calcium channel blockers ers for the practising anaesthetist. These drugs have Traditional played an expanding role in therapeutics since their Angina pectoris introduction and thus anaesthetists can expect to see Arrhythmias increasing numbers of patients presenting for anaes- Hypertension thesia who are being treated with calcium channel Newer and investigational Cardiac blockers. Other reviews have emphasized the basic - Hypertrophic cardiomyopathy pharmacology of calcium channel blockers. 1-7 - Cold cardioplegia - Pulmonary hypertension Physiology - calcium/calcium channel blockers Actions on platelets Calcium plays an important role in many physio- Asthma Obstetrics logical processes, such as blood coagulation, en- - Premature labor zyme systems, muscle contraction, bone metabo- - Pre-eclampsia lism, synaptic transmission, and cell membrane Achalasia and oesophageal spasm excitability. Especially important is the role of Increased intraocular pressure therapy calcium in myocardial contractility and conduction Protective effect on kidney after radiocontrast Cerebral vasospasm as well as in vascular smooth muscle reactivity. 7 Induced hypotensive anaesthesia Thus, it can be anticipated that any drug interfering Drag interactions with calcium channel blockers with the action of calcium could have widespread With anaesthetic agents effects. Inhalation agents In order to understand the importance of calcium - Effect on haemodynamics - Effect on MAC in cellular excitation, it is necessary to review some Neuromuscular blockers membrane physiology. Cell membranes are pri- Effects on epinephrine-induced arrhythmias marily phospholipids arranged in a bilayer. -

Conlaining Lidocaine Or Benzocaine Mark E
Anesth Prog 35:9-13 1988 Antimicrobial Properties of Topical Anestletc Liquids Conlaining Lidocaine or Benzocaine Mark E. Morrow, DDS, MSD,* and Charles W. Berry, PhDt *Department of Pediatric Dentistry, tDepartment of Microbiology, Baylor College of Dentistry, Dallas, Texas Six species of microorganisms commonly found ganisms have been isolated from the oral cavity,3 it is within the oral cavity were exposed for either one logical that an antiseptic agent should be wiped on the minute or two hours to 5% lidocaine liquid mucosa before the administration of an injectable local topical anesthetic and benzocaine liquid topical anesthetic in the dental setting. However, few dentists anesthetic. Mixtures of microorganisms and currently follow this regimen.4 Instead, the use of a anesthetics were diluted and plated onto a brain topical anesthetic just before administration of a local heart infusion medium. Reduction in cell viability anesthetic to block the pain of the needle puncture is very was 73-100% after exposure to the anesthetic common.5 agents when compared with the saline/buffer The possible antimicrobial properties of local anesthe- controls. A significant reduction (p < .005) in cell tic agents were originally described by Jonnesco.6 He growth by Streptococcus mutans, S. sanguis, S. indicated that there was no need to sterilize local anesthe- mitis, S. salivarius, Actinomyces viscosus, and tics used for spinal anesthesia because of their antiseptic Candida albicans was associated with a one- characteristics. The earliest work documenting the in vitro minute and two-hour exposure to lidocaine, antimicrobial activity of anesthetic agents was reported by benzocaine, 5% lidocaine, and the benzocaine Schlegal and Swan,7 who described the inhibition of vehicle control. -

Pharmacological Treatments Protocols of Alcohol and Drugs Abuse
Pharmacological Treatments Protocols of Alcohol and Drugs Abuse 1 Purpose of the protocols: Use and abuse of drugs and alcohol is becoming common and can have serious and harmful consequences on individuals, families, and society. Care with a tailored treatment program and follow-up options can be crucial to success. Treatment should include both medical and mental health services as needed in managing withdrawal symptoms, prevent relapse, and treat co- occurring conditions. Follow-up care may include community- or family-based recovery support systems. These protocols have been developed to guide medical practitioners and nurses in the use of the most effective available treatments of alcohol and drug abuse in the in-patient and out- patient settings and serve as a framework for clinical decisions and supporting best practices. Targeted end users: • Psychiatry and Addiction Medicine Consultants, Specialists and Residents • Nurses • Psychiatry clinical pharmacists • Pharmacists 2 TABLE OF CONTENTS 1. Chapter (1) Alcohol 4 3.1 Introduction 4 3.2 Intoxication 4 3.3 Withdrawal 7 2. Chapter (2) Benzodiazepines 21 2.1 Introduction 21 2.2 Intoxication 21 2.3 Withdrawal 22 3. Chapter (3) Opioids 27 3.1 Introduction 27 3.2 Intoxication 28 3.3 Withdrawal 29 4. Chapter (4) Psychostimulants 38 2.1 Introduction 38 2.2 Intoxication 39 2.3 Withdrawal 40 5. Chapter (5) Cannabis 41 3.1 Introduction 41 3.2 Intoxication 41 3.3 Withdrawal 43 6. References 46 3 CHAPTER 1 ALCOHOL INTRODUCTION Alcohol is a Central Nervous System (CNS) depressant. Its’ psychoactive properties contribute to changes in mood, cognition and behavior. -

(Lidocaine Hcl Injection, USP) 3 Xylocaine
1 721566-04 451175A/Revised February 2010 ® 2 Xylocaine (lidocaine HCl Injection, USP) ® 3 Xylocaine (lidocaine HCl and epinephrine Injection, USP) 4 For Infiltration and Nerve Block 5 Rx only 6 DESCRIPTION : 7 Xylocaine (lidocaine HCl) Injections are sterile, nonpyrogenic, aqueous solutions that contain a 8 local anesthetic agent with or without epinephrine and are administered parenterally by injection. 9 See INDICATIONS for specific uses. 10 Xylocaine solutions contain lidocaine HCl, which is chemically designated as acetamide, 11 2-(diethylamino)-N-(2,6-dimethylphenyl)-, monohydrochloride and has the molecular wt. 270.8. 12 Lidocaine HCl (C14H22N2O • HCl) has the following structural formula: 13 14 15 Epinephrine is (-) -3, 4-Dihydroxy-α-[(methylamino) methyl] benzyl alcohol and has the 16 molecular wt. 183.21. Epinephrine (C9H13NO3) has the following structural formula: 17 18 Dosage forms listed as Xylocaine-MPF indicate single dose solutions that are Methyl 19 Paraben Free (MPF). 20 Xylocaine MPF is a sterile, nonpyrogenic, isotonic solution containing sodium chloride. 21 Xylocaine in multiple dose vials: Each mL also contains 1 mg methylparaben as antiseptic 22 preservative. The pH of these solutions is adjusted to approximately 6.5 (5.0–7.0) with sodium 23 hydroxide and/or hydrochloric acid. 1 1 Xylocaine MPF with Epinephrine is a sterile, nonpyrogenic, isotonic solution containing 2 sodium chloride. Each mL contains lidocaine hydrochloride and epinephrine, with 0.5 mg 3 sodium metabisulfite as an antioxidant and 0.2 mg citric acid as a stabilizer. Xylocaine with 4 Epinephrine in multiple dose vials: Each mL also contains 1 mg methylparaben as antiseptic 5 preservative. -

World Health Organization Model List of Essential Medicines, 21St List, 2019
World Health Organizatio n Model List of Essential Medicines 21st List 2019 World Health Organizatio n Model List of Essential Medicines 21st List 2019 WHO/MVP/EMP/IAU/2019.06 © World Health Organization 2019 Some rights reserved. This work is available under the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 IGO licence (CC BY-NC-SA 3.0 IGO; https://creativecommons.org/licenses/by-nc-sa/3.0/igo). Under the terms of this licence, you may copy, redistribute and adapt the work for non-commercial purposes, provided the work is appropriately cited, as indicated below. In any use of this work, there should be no suggestion that WHO endorses any specific organization, products or services. The use of the WHO logo is not permitted. If you adapt the work, then you must license your work under the same or equivalent Creative Commons licence. If you create a translation of this work, you should add the following disclaimer along with the suggested citation: “This translation was not created by the World Health Organization (WHO). WHO is not responsible for the content or accuracy of this translation. The original English edition shall be the binding and authentic edition”. Any mediation relating to disputes arising under the licence shall be conducted in accordance with the mediation rules of the World Intellectual Property Organization. Suggested citation. World Health Organization Model List of Essential Medicines, 21st List, 2019. Geneva: World Health Organization; 2019. Licence: CC BY-NC-SA 3.0 IGO. Cataloguing-in-Publication (CIP) data. CIP data are available at http://apps.who.int/iris. -

Lidocaine Hcl) Solution
451424/Issued: September 2014 ® 2% Xylocaine Viscous (lidocaine HCl) Solution A Topical Anesthetic for the Mucous Membranes of the Mouth and Pharynx. WARNING: Life-threatening and fatal events in infants and young children Postmarketing cases of seizures, cardiopulmonary arrest, and death in patients under the age of 3 years have been reported with use of Xylocaine 2% Viscous Solution when it was not administered in strict adherence to the dosing and administration recommendations. In the setting of teething pain, Xylocaine 2% Viscous Solution should generally not be used. For other conditions, the use of the product in patients less than 3 years of age should be limited to those situations where safer alternatives are not available or have been tried but failed. To decrease the risk of serious adverse events with use of Xylocaine 2% Viscous Solution, instruct caregivers to strictly adhere to the prescribed dose and frequency of administration and store the prescription bottle safely out of reach of children. DESCRIPTION: Xylocaine (lidocaine HCl) 2% Viscous Solution contains a local anesthetic agent and is administered topically. Xylocaine 2% Viscous Solution contains lidocaine HCl, which is chemically designated as acetamide, 2-(diethylamino)-N-(2,6-dimethylphenyl)-, monohydrochloride and has the following structural formula: 1 Reference ID: 3633341 The molecular formula of lidocaine is C14H22N2O. The molecular weight is 234.34. COMPOSITION OF SOLUTION: Each mL contains 20 mg of lidocaine HCl, flavoring, saccharin sodium, methylparaben, propylparaben and sodium carboxymethylcellulose in purified water. The pH is adjusted to 6.0 to 7.0 with sodium hydroxide. CLINICAL PHARMACOLOGY Mechanism of Action Lidocaine stabilizes the neuronal membrane by inhibiting the ionic fluxes required for the initiation and conduction of impulses, thereby effecting local anesthetic action. -

Diltiazem Added to Local Anesthetic in Sonar Guided Coracoid Infraclavicular Brachial Plexus Block
Journal of Clinical Anesthesia and Pain Medicine Research Article Diltiazem Added to Local Anesthetic in Sonar Guided Coracoid Infraclavicular Brachial Plexus Block. Dose Sparing Effect This article was published in the following Scient Open Access Journal: Journal of Clinical Anesthesia and Pain Medicine Received November 01, 2017; Accepted March 15, 2018; Published April 07, 2018 Abstract 1 1 Ashraf E Alzeftawy *, Hesham E Elsheikh Background: This study was performed to evaluate the addition of diltiazem to local 1Assistant Professors of Anesthesia and Surgical anesthetic during sonar guided infraclavicular brachial plexus block as regards to its intra Intensive care, Department of Anesthesia and and postoperative analgesic quality. Surgical Intensive care, Tanta University, Tanta, Egypt Methods: In a randomized, prospective double blind study, 90 patients aged between 18-70 years with ASA Grade I and II, were randomly classified into 3 groups. Group I received 30 ml containing 14 ml 0.5% bupivacaine and 14 ml lidocaine 2% plus 2 ml saline and group II received same volume of local anesthetic and diltiazem 20mg in 2 ml saline. while group III received 30 ml containing 10 ml 0.5% bupivacaine , 10 ml lidocaine 2% and diltiazem 20mg in 10 ml saline. Onset of sensory and motor block , quality of the block , postoperative analgesia, and any complications of the procedure were recorded. Results: Onset of sensory and motor block was significantly shorter in group II compared to other groups. Quality of the block in the three groups was comparable. Significantly prolonged duration of sensory and motor block and prolonged duration of analgesia were present in group II compared to other groups. -

Children's Preference of Benzocaine Gel Versus the Lidocaine Patch
Scientific Article Children’s Preference of Benzocaine Gel Versus the Lidocaine Patch Sonia J. Wu, DMD Kell Julliard, MA, MFA Dr. Wu is a former resident, Pediatric Dental Department, and Mr. Julliard is research program director, Lutheran Medical Center, Brooklyn, NY. Correspond with Mr. Julliard at [email protected] Abstract Purpose: This study compared pain, acceptance, and preference associated with 2 topi- cal anesthetics: benzocaine gel and lidocaine patch (DentiPatch). Methods: Thirty patients aged 3 to 10 years participated in this within-subjects study. All children required identical or similar dental work bilaterally (restorations, extractions, endodontic procedures, or sealants). Subjects chose either DentiPatch or benzocaine gel at the first visit. The anesthetic the child did not choose was used at the second visit. The Whali-Wong scale was used to measure comfort before and after application of topi- cal anesthetic and after injection, and the Sounds, Eyes, Motor (SEM) scale measured pain upon injection. Results: At the first visit, 80% of subjects selected DentiPatch; 60% of subjects made their choice based on appearance. Younger children more than older children were in- fluenced by appearance in their selection. After trying both topical anesthetics, 77% preferred DentiPatch; final preference and either age or gender were not significantly related. The gel had greater scores than the patch for the Sounds pain value and for the SEM scale composite score. Conclusions: The lidocaine patch was associated with some objective evidence of re- duced pain compared to the gel and was preferred by most children.(Pediatr Dent. 2003;25:401-405) KEYWORDS: TOPICAL ANESTHETIC, BENZOCAINE GEL, LIDOCAINE PATCH, DENTIPATCH Received July 1, 2002 Revision Accepted February 6, 2003 ehavior management in pediatric dentistry encom- thesia and has unit-dose convenience.