Fenestrations Control Resting-State Block of a Voltage-Gated Sodium
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The National Drugs List
^ ^ ^ ^ ^[ ^ The National Drugs List Of Syrian Arab Republic Sexth Edition 2006 ! " # "$ % &'() " # * +$, -. / & 0 /+12 3 4" 5 "$ . "$ 67"5,) 0 " /! !2 4? @ % 88 9 3: " # "$ ;+<=2 – G# H H2 I) – 6( – 65 : A B C "5 : , D )* . J!* HK"3 H"$ T ) 4 B K<) +$ LMA N O 3 4P<B &Q / RS ) H< C4VH /430 / 1988 V W* < C A GQ ") 4V / 1000 / C4VH /820 / 2001 V XX K<# C ,V /500 / 1992 V "!X V /946 / 2004 V Z < C V /914 / 2003 V ) < ] +$, [2 / ,) @# @ S%Q2 J"= [ &<\ @ +$ LMA 1 O \ . S X '( ^ & M_ `AB @ &' 3 4" + @ V= 4 )\ " : N " # "$ 6 ) G" 3Q + a C G /<"B d3: C K7 e , fM 4 Q b"$ " < $\ c"7: 5) G . HHH3Q J # Hg ' V"h 6< G* H5 !" # $%" & $' ,* ( )* + 2 ا اوا ادو +% 5 j 2 i1 6 B J' 6<X " 6"[ i2 "$ "< * i3 10 6 i4 11 6! ^ i5 13 6<X "!# * i6 15 7 G!, 6 - k 24"$d dl ?K V *4V h 63[46 ' i8 19 Adl 20 "( 2 i9 20 G Q) 6 i10 20 a 6 m[, 6 i11 21 ?K V $n i12 21 "% * i13 23 b+ 6 i14 23 oe C * i15 24 !, 2 6\ i16 25 C V pq * i17 26 ( S 6) 1, ++ &"r i19 3 +% 27 G 6 ""% i19 28 ^ Ks 2 i20 31 % Ks 2 i21 32 s * i22 35 " " * i23 37 "$ * i24 38 6" i25 39 V t h Gu* v!* 2 i26 39 ( 2 i27 40 B w< Ks 2 i28 40 d C &"r i29 42 "' 6 i30 42 " * i31 42 ":< * i32 5 ./ 0" -33 4 : ANAESTHETICS $ 1 2 -1 :GENERAL ANAESTHETICS AND OXYGEN 4 $1 2 2- ATRACURIUM BESYLATE DROPERIDOL ETHER FENTANYL HALOTHANE ISOFLURANE KETAMINE HCL NITROUS OXIDE OXYGEN PROPOFOL REMIFENTANIL SEVOFLURANE SUFENTANIL THIOPENTAL :LOCAL ANAESTHETICS !67$1 2 -5 AMYLEINE HCL=AMYLOCAINE ARTICAINE BENZOCAINE BUPIVACAINE CINCHOCAINE LIDOCAINE MEPIVACAINE OXETHAZAINE PRAMOXINE PRILOCAINE PREOPERATIVE MEDICATION & SEDATION FOR 9*: ;< " 2 -8 : : SHORT -TERM PROCEDURES ATROPINE DIAZEPAM INJ. -

Hygroscopicity of Pharmaceutical Crystals
HYGROSCOPICITY OF PHARMACEUTICAL CRYSTALS A DISSERTATION SUBMITTED TO THE FACULTY OF GRADUATE SCHOOL OF THE UNIVERSITY OF MINNESOTA BY DABING CHEN IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY RAJ SURYANARAYANAN (ADVISER) JANUARY, 2009 © Dabing Chen, January / 2009 ACKNOWLEDGEMENTS I am very grateful to my thesis advisor, Prof. Raj Suryanarayanan, for his constant guidance, support, and encouragement throughout my research. Without his help, the completion of this thesis would be impossible. His friendship and advices are precious to my professional and personal growth and will help me overcome many difficulties in my future career. I would like to take the opportunity to thank Prof. David J.W. Grant, who was my advisor during the first three years in graduate school and led me into the research area of physical pharmacy. It was my great honor to have worked for him, and he will always live as a role model in my life. Many thanks to Dr. Zheng Jane Li at Boehringer Ingelheim Pharmaceuticals (BI) for her invaluable advice as an industrial mentor and also for agreeing to serve on my committee. I sincerely appreciate her helpful discussions, revision of the manuscripts, and supervision of my research. I also want to thank her for providing me the internship opportunity at BI. I thank Dr. Timothy S. Wiedmann and Dr. Theodore P. Labuza for serving on my committee and for critically reviewing my thesis. I also want to thank Dr. Timothy S. Wiedmann for allowing me the use of the HPLC instruments in his lab and also for his advice as the Director of Graduate Studies. -

Topical Therapy As a Treatmentfor Brachioradial
Journal of Case Reports: Open Access Case report Open Access Topical Therapy as a Treatmentfor Brachioradial Pruritis: a Case Report Brianna De Souza M.D, Amy McMichael M.D* Department of Dermatology, Wake Forest University School of Medicine, Winston-Salem, North Carolina *Corresponding author: Amy McMichael, MD Department of Dermatology, Wake Forest Baptist Medical Center,1 Medical Center Blvd, Winston-Salem, NC 27157, Phone: 336-716-7882, Email: [email protected] Received Date: April 12, 2019 Accepted Date: May 06, 2019 Published Date: May 08, 2019 Citation: Brianna De Souza (2019) Topical Therapy as a Treatmentfor Brachioradial Pruritis: a Case Report. Case Reports: Open Access 4: 1-5. Abstract Management of brachioradial pruritus (BRP) presents a formidable challenge to dermatologists and neurologists. BRP is a rare, neurocutaneous condition characterized by sharply localized, chronic pain with associated itching, burning, stinging, and or tingling sensation. Effective care of this patient population is confounded by limitations within the litera- ture, comprised of case series and case reports. We present a case of one middle-aged female with a chronic history of BRP recalcitrant to the following oral therapies: pregabalin, gabapentin, mirtazapine, prednisone, and amitriptyline, as well as topical triamcinolone. After being evaluated in the clinic, the patient was started on combination therapy withKetamine 10%, Amitriptyline 5%, and Lidocaine 5% topical cream to which she responded. Keywords: Brachioradial pruritus, Brachioradial, Pruritus, Neurocutaneous ©2019 The Authors. Published by the JScholar under the terms of the Crea- tive Commons Attribution License http://creativecommons.org/licenses/ by/3.0/, which permits unrestricted use, provided the original author and source are credited. -

F 009 035 Benzocaine 20%, Lidocaine Hydrochoride 7
MEDISCA® NETWORK INC. TECHNICAL SUPPORT SERVICES FORMULATION CHEMISTRY DEPARTMENT TOLL-FREE: 866-333-7811 TELEPHONE: 514-905-5096 FAX: 514-905-5097 [email protected] 1/4/2021; Page 1 Suggested Benzocaine 20%, Lidocaine Hydrochloride 7%, Tetracaine Hydrochloride 7% FIN F 009 035 Formula Topical Gel (Suspension, 30 g) SUGGESTED FORMULATION Lot Expiry Ingredient Listing Qty. Unit NDC # Supplier Number Date Benzocaine, USP 6.000 g Lidocaine Hydrochloride, USP TBD Tetracaine Hydrochloride, USP 2.100 g Polysorbate 80, NF 0.5 mL Ethoxy Diglycol, NF 0.5 mL Medisca VersaPro™ Anhydrous Base 1.50 g Medisca VersaPro™ Anhydrous Base TBD MEDISCA® NETWORK INC. TECHNICAL SUPPORT SERVICES FORMULATION CHEMISTRY DEPARTMENT TOLL-FREE: 866-333-7811 TELEPHONE: 514-905-5096 FAX: 514-905-5097 [email protected] 1/4/2021; Page 2 Suggested Benzocaine 20%, Lidocaine Hydrochloride 7%, Tetracaine Hydrochloride 7% FIN F 009 035 Formula Topical Gel (Suspension, 30 g) SPECIAL PREPARATORY CONSIDERATIONS Ingredient-Specific Information Benzocaine, Tetracaine Hydrochloride, Polysorbate Light Sensitive (protect from light whenever possible): 80 Tetracaine Hydrochloride, Polysorbate 80, Ethoxy Hygroscopic (protect from moisture whenever possible): Diglycol Oxygen Sensitive (protect from air whenever possible): Polysorbate 80 Narrow Therapeutic Index Lidocaine Hydrochloride Suggested Preparatory Guidelines ■ Non-Sterile Preparation □ Sterile Preparation Processing Error / To account for processing error considerations during preparation, it is suggested to Testing Considerations: measure an additional 12 to 15% of the required quantities of ingredients. Special Instruction: This formula may contain one or more Active Pharmaceutical Ingredients (APIs) that may be classified as hazardous, please refer & verify the current NIOSH list of Antineoplastic and Other Hazardous Drugs in Healthcare Settings. -

Antagonism of Lidocaine Inhibition by Open-Channel Blockers That Generate Resurgent Na Current
4976 • The Journal of Neuroscience, March 13, 2013 • 33(11):4976–4987 Cellular/Molecular Antagonism of Lidocaine Inhibition by Open-Channel Blockers That Generate Resurgent Na Current Jason S. Bant,1,3 Teresa K. Aman,2,3 and Indira M. Raman1,2,3 1Interdepartmental Biological Sciences Program, 2Northwestern University Interdepartmental Neuroscience Program, and 3Department of Neurobiology, Northwestern University, Evanston, Illinois 60208 Na channels that generate resurgent current express an intracellular endogenous open-channel blocking protein, whose rapid binding upon depolarization and unbinding upon repolarization minimizes fast and slow inactivation. Na channels also bind exogenous com- pounds, such as lidocaine, which functionally stabilize inactivation. Like the endogenous blocking protein, these use-dependent inhibi- tors bind most effectively at depolarized potentials, raising the question of how lidocaine-like compounds affect neurons with resurgent Na current. We therefore recorded lidocaine inhibition of voltage-clamped, tetrodotoxin-sensitive Na currents in mouse Purkinje neu- rons, which express a native blocking protein, and in mouse hippocampal CA3 pyramidal neurons with and without a peptide from the   cytoplasmic tail of NaV 4 (the 4 peptide), which mimics endogenous open-channel block. To control channel states during drug exposure, lidocaine was applied with rapid-solution exchange techniques during steps to specific voltages. Inhibition of Na currents by lidocaine was diminished by either the 4 peptide or the native blocking protein. In peptide-free CA3 cells, prolonging channel opening with a site-3 toxin, anemone toxin II, reduced lidocaine inhibition; this effect was largely occluded by open-channel blockers, suggesting that lidocaine binding is favored by inactivation but prevented by open-channel block. -
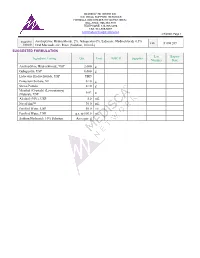
Amitriptyline Hydrochloride 2%, Gabapentin 6%, Lidocaine Hydrochloride 0.5% FIN F 008 269 Formula Oral Mucoadhesive Rinse (Solution, 100 Ml)
MEDISCA® NETWORK INC. TECHNICAL SUPPORT SERVICES FORMULATION CHEMISTRY DEPARTMENT TOLL-FREE: 866-333-7811 TELEPHONE: 514-905-5096 FAX: 514-905-5097 [email protected] 4/7/2020; Page 1 Suggested Amitriptyline Hydrochloride 2%, Gabapentin 6%, Lidocaine Hydrochloride 0.5% FIN F 008 269 Formula Oral Mucoadhesive Rinse (Solution, 100 mL) SUGGESTED FORMULATION Lot Expiry Ingredient Listing Qty. Unit NDC # Supplier Number Date Amitriptyline Hydrochloride, USP 2.000 g Gabapentin, USP 6.000 g Lidocaine Hydrochloride, USP TBD Potassium Sorbate, NF 0.10 g Stevia Powder 0.10 g Menthol (Crystals) (Levorotatory) 0.02 g (Natural), USP Alcohol (95%), USP 5.0 mL NovaFilm™ 30.0 mL Purified Water, USP 50.0 mL Purified Water, USP q.s. to 100.0 mL Sodium Hydroxide 10% Solution As required MEDISCA® NETWORK INC. TECHNICAL SUPPORT SERVICES FORMULATION CHEMISTRY DEPARTMENT TOLL-FREE: 866-333-7811 TELEPHONE: 514-905-5096 FAX: 514-905-5097 [email protected] 4/7/2020; Page 2 Suggested Amitriptyline Hydrochloride 2%, Gabapentin 6%, Lidocaine Hydrochloride 0.5% FIN F 008 269 Formula Oral Mucoadhesive Rinse (Solution, 100 mL) SPECIAL PREPARATORY CONSIDERATIONS Ingredient-Specific Information Light Sensitive (protect from light whenever possible): Amitriptyline Hydrochloride, Gabapentin Hygroscopic (protect from moisture whenever possible): Stevia Powder Narrow Therapeutic Index Lidocaine Hydrochloride Suggested Preparatory Guidelines ■ Non-Sterile Preparation □ Sterile Preparation Processing Error / To account for processing error and pH testing considerations during preparation, it is Testing Considerations: suggested to measure an additional 3 to 5% of the required quantities of ingredients. Special Instruction: This formula may contain one or more Active Pharmaceutical Ingredients (APIs) that may be classified as hazardous, please refer & verify the current NIOSH list of Antineoplastic and Other Hazardous Drugs in Healthcare Settings, 2016. -

The Effects of Lidocaine and Mefenamic Acid on Post-Episiotomy
Shiraz E-Med J. 2016 March; 17(3):e36286. doi: 10.17795/semj36286. Published online 2016 March 27. Research Article The Effects of Lidocaine and Mefenamic Acid on Post-Episiotomy Pain: A Comparative Study Masoumeh Delaram,1,* Lobat Jafar Zadeh,2 and Sahand Shams3 1Faculty of Nursing and Midwifery, Shahrekord University of Medical Sciences, Shahrekord, IR Iran 2Faculty of Medicine, Shahrekord University of Medical Sciences, Shahrekord, IR Iran 3Faculty of Veterinary Medicine, Shahrekord University, Shahrekord, IR Iran *Corresponding author: Masoumeh Delaram, Faculty of Nursing and Midwifery, Shahrekord University of Medical Sciences, Shahrekord, IR Iran. Tel: +98-3813335648, Fax: +98-3813346714, E-mail: [email protected] Received 2016 January 13; Revised 2016 February 29; Accepted 2016 March 04. Abstract Background: Most women suffer pain following an episiotomy and oral non-steroidal anti-inflammatory drugs are commonly used for pain relief. Due to the gastrointestinal side effects of oral drugs, it seems that women are more accepting of topical medications for pain relief. Objectives: Therefore, the aim of this study was to compare the effects of lidocaine and mefenamic acid on post-episiotomy pain. Patients and Methods: This clinical trial was carried out in 2011. It involved sixty women with singleton pregnancy who were given an episiotomy at 38 to 42 weeks of gestation. The participants were randomly divided into two groups. One group received 2% lido- caine cream (n = 30), while the other group received 250 mg of mefenamic acid (n = 30). The data were collected via a questionnaire and a visual analog scale. Pain intensity was compared from the first complaint by the mother and at 6, 12, and 24 hours after the delivery in both groups. -

Adverse Effects of Medications on Oral Health
Adverse Effects of Medications on Oral Health Dr. James Krebs, BS Pharm, MS, PharmD Director of Experiential Education College of Pharmacy, University of New England Presented by: Rachel Foster PharmD Candidate, Class of 2014 University of New England October 2013 Objectives • Describe the pathophysiology of various medication-related oral reactions • Recognize the signs and symptoms associated with medication-related oral reactions • Identify the populations associated with various offending agents • Compare the treatment options for medication-related oral reactions Medication-related Oral Reactions • Stomatitis • Oral Candidiasis • Burning mouth • Gingival hyperplasia syndrome • Alterations in • Glossitis salivation • Erythema • Alterations in taste Multiforme • Halitosis • Oral pigmentation • Angioedema • Tooth discoloration • Black hairy tongue Medication-related Stomatitis • Clinical presentation – Aphthous-like ulcers, mucositis, fixed-drug eruption, lichen planus1,2 – Open sores in the mouth • Tongue, gum line, buccal membrane – Patient complaint of soreness or burning http://www.virtualmedicalcentre.com/diseases/oral-mucositis-om/92 0 http://www.virtualmedicalcentre.com/diseases/oral-mucositis-om/920 Medication-related Stomatitis • Offending agents1,2 Medication Indication Patient Population Aspirin •Heart health • >18 years old •Pain reliever • Cardiac patients NSAIDs (i.e. Ibuprofen, •Headache General population naproxen) •Pain reliever •Fever reducer Chemotherapy (i.e. •Breast cancer •Oncology patients methotrexate, 5FU, •Colon -

Medication Guide for a Safe Recovery
Medication Guide For A Safe Recovery A guide to maintaining sobriety while receiving treatment for other health problems. Revision 1.0 -April 2008 Table of Contents Introduction..................................................................................2 How to Use this Guide..................................................................3 Class A Drugs (Absolutely Avoid)................................................4 Class B Drugs................................................................................8 (With Addiction Medicine Specialist/Doctor Approval Only) Class C Drugs (Generally Safe to Take).....................................12 Alcohol-Free Products..................................................................16 Incidental Exposure Index...........................................................22 www.talbottcampus.com Introduction From the Talbott Recovery Campus Welcome to the Talbott Recovery Campus guide for a safe and sustained recovery. This document was developed through a collaborative effort between some of the best minds in addiction care today and will help you make wise decisions, ensuring that medications you may be prescribed and incidental exposure to alcohol do not threaten your hard won recovery. This guide is divided into three sections and is based on the drug classification system developed nearly 20 years ago by Dr. Paul Earley and recently expanded on by Bruce Merkin, M.D., Renee Enstrom, Nicholas Link and the staff at Glenbeigh hospital. Part one provides a way of categorizing medications -

Anaesthetic Implications of Calcium Channel Blockers
436 Anaesthetic implications of calcium channel Leonard C. Jenkins aA MD CM FRCPC blockers Peter J. Scoates a sc MD FRCPC CONTENTS The object of this review is to emphasize the anaesthetic implications of calcium channel block- Physiology - calcium/calcium channel blockers Uses of calcium channel blockers ers for the practising anaesthetist. These drugs have Traditional played an expanding role in therapeutics since their Angina pectoris introduction and thus anaesthetists can expect to see Arrhythmias increasing numbers of patients presenting for anaes- Hypertension thesia who are being treated with calcium channel Newer and investigational Cardiac blockers. Other reviews have emphasized the basic - Hypertrophic cardiomyopathy pharmacology of calcium channel blockers. 1-7 - Cold cardioplegia - Pulmonary hypertension Physiology - calcium/calcium channel blockers Actions on platelets Calcium plays an important role in many physio- Asthma Obstetrics logical processes, such as blood coagulation, en- - Premature labor zyme systems, muscle contraction, bone metabo- - Pre-eclampsia lism, synaptic transmission, and cell membrane Achalasia and oesophageal spasm excitability. Especially important is the role of Increased intraocular pressure therapy calcium in myocardial contractility and conduction Protective effect on kidney after radiocontrast Cerebral vasospasm as well as in vascular smooth muscle reactivity. 7 Induced hypotensive anaesthesia Thus, it can be anticipated that any drug interfering Drag interactions with calcium channel blockers with the action of calcium could have widespread With anaesthetic agents effects. Inhalation agents In order to understand the importance of calcium - Effect on haemodynamics - Effect on MAC in cellular excitation, it is necessary to review some Neuromuscular blockers membrane physiology. Cell membranes are pri- Effects on epinephrine-induced arrhythmias marily phospholipids arranged in a bilayer. -

Conlaining Lidocaine Or Benzocaine Mark E
Anesth Prog 35:9-13 1988 Antimicrobial Properties of Topical Anestletc Liquids Conlaining Lidocaine or Benzocaine Mark E. Morrow, DDS, MSD,* and Charles W. Berry, PhDt *Department of Pediatric Dentistry, tDepartment of Microbiology, Baylor College of Dentistry, Dallas, Texas Six species of microorganisms commonly found ganisms have been isolated from the oral cavity,3 it is within the oral cavity were exposed for either one logical that an antiseptic agent should be wiped on the minute or two hours to 5% lidocaine liquid mucosa before the administration of an injectable local topical anesthetic and benzocaine liquid topical anesthetic in the dental setting. However, few dentists anesthetic. Mixtures of microorganisms and currently follow this regimen.4 Instead, the use of a anesthetics were diluted and plated onto a brain topical anesthetic just before administration of a local heart infusion medium. Reduction in cell viability anesthetic to block the pain of the needle puncture is very was 73-100% after exposure to the anesthetic common.5 agents when compared with the saline/buffer The possible antimicrobial properties of local anesthe- controls. A significant reduction (p < .005) in cell tic agents were originally described by Jonnesco.6 He growth by Streptococcus mutans, S. sanguis, S. indicated that there was no need to sterilize local anesthe- mitis, S. salivarius, Actinomyces viscosus, and tics used for spinal anesthesia because of their antiseptic Candida albicans was associated with a one- characteristics. The earliest work documenting the in vitro minute and two-hour exposure to lidocaine, antimicrobial activity of anesthetic agents was reported by benzocaine, 5% lidocaine, and the benzocaine Schlegal and Swan,7 who described the inhibition of vehicle control. -

Pharmaceuticals As Environmental Contaminants
PharmaceuticalsPharmaceuticals asas EnvironmentalEnvironmental Contaminants:Contaminants: anan OverviewOverview ofof thethe ScienceScience Christian G. Daughton, Ph.D. Chief, Environmental Chemistry Branch Environmental Sciences Division National Exposure Research Laboratory Office of Research and Development Environmental Protection Agency Las Vegas, Nevada 89119 [email protected] Office of Research and Development National Exposure Research Laboratory, Environmental Sciences Division, Las Vegas, Nevada Why and how do drugs contaminate the environment? What might it all mean? How do we prevent it? Office of Research and Development National Exposure Research Laboratory, Environmental Sciences Division, Las Vegas, Nevada This talk presents only a cursory overview of some of the many science issues surrounding the topic of pharmaceuticals as environmental contaminants Office of Research and Development National Exposure Research Laboratory, Environmental Sciences Division, Las Vegas, Nevada A Clarification We sometimes loosely (but incorrectly) refer to drugs, medicines, medications, or pharmaceuticals as being the substances that contaminant the environment. The actual environmental contaminants, however, are the active pharmaceutical ingredients – APIs. These terms are all often used interchangeably Office of Research and Development National Exposure Research Laboratory, Environmental Sciences Division, Las Vegas, Nevada Office of Research and Development Available: http://www.epa.gov/nerlesd1/chemistry/pharma/image/drawing.pdfNational