Seprase; an Overview of an Important Matrix Serine Protease
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

(12) Patent Application Publication (10) Pub. No.: US 2016/0346364 A1 BRUNS Et Al
US 2016.0346364A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2016/0346364 A1 BRUNS et al. (43) Pub. Date: Dec. 1, 2016 (54) MEDICAMENT AND METHOD FOR (52) U.S. Cl. TREATING INNATE IMMUNE RESPONSE CPC ........... A61K 38/488 (2013.01); A61K 38/482 DISEASES (2013.01); C12Y 304/23019 (2013.01); C12Y 304/21026 (2013.01); C12Y 304/23018 (71) Applicant: DSM IPASSETS B.V., Heerlen (NL) (2013.01); A61K 9/0053 (2013.01); C12N 9/62 (2013.01); A23L 29/06 (2016.08); A2ID 8/042 (72) Inventors: Maaike Johanna BRUINS, Kaiseraugst (2013.01); A23L 5/25 (2016.08); A23V (CH); Luppo EDENS, Kaiseraugst 2002/00 (2013.01) (CH); Lenneke NAN, Kaiseraugst (CH) (57) ABSTRACT (21) Appl. No.: 15/101,630 This invention relates to a medicament or a dietary Supple (22) PCT Filed: Dec. 11, 2014 ment comprising the Aspergillus niger aspergilloglutamic peptidase that is capable of hydrolyzing plant food allergens, (86). PCT No.: PCT/EP2014/077355 and more particularly, alpha-amylase/trypsin inhibitors, thereby treating diseases due to an innate immune response S 371 (c)(1), in humans, and/or allowing to delay the onset of said (2) Date: Jun. 3, 2016 diseases. The present invention relates to the discovery that (30) Foreign Application Priority Data the Aspergillus niger aspergilloglutamic peptidase is capable of hydrolyzing alpha-amylase/trypsin inhibitors that are Dec. 11, 2013 (EP) .................................. 13196580.8 present in wheat and related cereals said inhibitors being strong inducers of innate immune response. Furthermore, Publication Classification the present invention relates to a method for hydrolyzing alpha-amylase/trypsin inhibitors comprising incubating a (51) Int. -

Clinical Study High Complement Factor I Activity in the Plasma of Children with Autism Spectrum Disorders
Hindawi Publishing Corporation Autism Research and Treatment Volume 2012, Article ID 868576, 6 pages doi:10.1155/2012/868576 Clinical Study High Complement Factor I Activity in the Plasma of Children with Autism Spectrum Disorders Naghi Momeni,1 Lars Brudin,2 Fatemeh Behnia,3 Berit Nordstrom,¨ 4 Ali Yosefi-Oudarji,5 Bengt Sivberg,4 Mohammad T. Joghataei,5 and Bengt L. Persson1 1 School of Natural Sciences, Linnaeus University, 39182 Kalmar, Sweden 2 Department of Clinical Physiology, Kalmar County Hospital, 39185 Kalmar, Sweden 3 Department of Occupational Therapy, University of Social Welfare and Rehabilitation Sciences, Tehran, Iran 4 Department of Health Sciences, Autism Research, Faculty of Medicine, Lund University, Box 157, 22100 Lund, Sweden 5 Cellular and Molecular Research Centre, Tehran University of Medical Sciences (TUMS), Tehran, Iran Correspondence should be addressed to Bengt Sivberg, [email protected] Received 17 June 2011; Revised 22 August 2011; Accepted 22 August 2011 Academic Editor: Judy Van de Water Copyright © 2012 Naghi Momeni et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Autism spectrum disorders (ASDs) are neurodevelopmental and behavioural syndromes affecting social orientation, behaviour, and communication that can be classified as developmental disorders. ASD is also associated with immune system abnormality. Im- mune system abnormalities may be caused partly by complement system factor I deficiency. Complement factor I is a serine pro- tease present in human plasma that is involved in the degradation of complement protein C3b, which is a major opsonin of the complement system. -

Mouse Prolyl Endopeptidase / PREP Protein (His Tag)
Mouse Prolyl endopeptidase / PREP Protein (His Tag) Catalog Number: 50288-M07B General Information SDS-PAGE: Gene Name Synonym: AI047692; AI450383; D10Wsu136e; PEP; Pop Protein Construction: A DNA sequence encoding the mature form of mouse PREP (Q9QUR6) (Leu2-Gln710) was expressed, with a polyhistidine tag at the N-terminus. Source: Mouse Expression Host: Baculovirus-Insect Cells QC Testing Purity: > 95 % as determined by SDS-PAGE Endotoxin: Protein Description < 1.0 EU per μg of the protein as determined by the LAL method Prolyl endopeptidase, also known as PREP, belongs to a distinct class of Stability: serine peptidases. It is a large cytosolic enzyme which was first described in the cytosol of rabbit brain as an oligopeptidase. Prolyl endopeptidase ℃ Samples are stable for up to twelve months from date of receipt at -70 degrades the nonapeptide bradykinin at the Pro-Phe bond. It is involved in the maturation and degradation of peptide hormones and neuropeptides His Predicted N terminal: such as alpha-melanocyte-stimulating hormone, luteinizing hormone- Molecular Mass: releasing hormone (LH-RH), thyrotropin-releasing hormone, angiotensin, neurotensin, oxytocin, substance P and vasopressin. Prolyl The recombinant mouse PREP consists of 727 amino acids and predicts a endopeptidase's activity is confined to action on oligopeptides of less than molecular mass of 82.9 KDa. It migrates as an approximately 79-83 KDa 10 kD and it has an absolute requirement for the trans-configuration of the band in SDS-PAGE under reducing conditions. peptide bond preceding proline. It cleaves peptide bonds at the C-terminal side of proline residues. Formulation: References Lyophilized from sterile 20mM Tris, 500mM NaCl, pH 7.4, 10% glycerol, 3mM DTT 1.Oliveira EB, et al. -

Effects of Glycosylation on the Enzymatic Activity and Mechanisms of Proteases
International Journal of Molecular Sciences Review Effects of Glycosylation on the Enzymatic Activity and Mechanisms of Proteases Peter Goettig Structural Biology Group, Faculty of Molecular Biology, University of Salzburg, Billrothstrasse 11, 5020 Salzburg, Austria; [email protected]; Tel.: +43-662-8044-7283; Fax: +43-662-8044-7209 Academic Editor: Cheorl-Ho Kim Received: 30 July 2016; Accepted: 10 November 2016; Published: 25 November 2016 Abstract: Posttranslational modifications are an important feature of most proteases in higher organisms, such as the conversion of inactive zymogens into active proteases. To date, little information is available on the role of glycosylation and functional implications for secreted proteases. Besides a stabilizing effect and protection against proteolysis, several proteases show a significant influence of glycosylation on the catalytic activity. Glycans can alter the substrate recognition, the specificity and binding affinity, as well as the turnover rates. However, there is currently no known general pattern, since glycosylation can have both stimulating and inhibiting effects on activity. Thus, a comparative analysis of individual cases with sufficient enzyme kinetic and structural data is a first approach to describe mechanistic principles that govern the effects of glycosylation on the function of proteases. The understanding of glycan functions becomes highly significant in proteomic and glycomic studies, which demonstrated that cancer-associated proteases, such as kallikrein-related peptidase 3, exhibit strongly altered glycosylation patterns in pathological cases. Such findings can contribute to a variety of future biomedical applications. Keywords: secreted protease; sequon; N-glycosylation; O-glycosylation; core glycan; enzyme kinetics; substrate recognition; flexible loops; Michaelis constant; turnover number 1. -

Reducing Immunoreactivity of Gliadins and Coeliac-Toxic Peptides Using Peptidases from L
catalysts Article Reducing Immunoreactivity of Gliadins and Coeliac-Toxic Peptides Using Peptidases from L. acidophilus 5e2 and A. niger Bartosz Brzozowski *, Katarzyna Stasiewicz, Mateusz Ostolski and Marek Adamczak Department of Food Biotechnology, Faculty of Food Sciences, University of Warmia and Mazury in Olsztyn, 10-718 Olsztyn, Poland; [email protected] (K.S.); [email protected] (M.O.); [email protected] (M.A.) * Correspondence: [email protected] Received: 21 July 2020; Accepted: 10 August 2020; Published: 11 August 2020 Abstract: Wheat storage proteins and products of their hydrolysis may cause coeliac sprue in genetically predisposed individuals with high expression of main histocompatibility complex HLA-DQ2 or DQ8, since by consuming wheat, they become exposed to proline- (P) and glutamine (Q)-rich gluten. In bread-making, the hydrolysis of gliadins and coeliac-toxic peptides occurs with varied efficiency depending on the fermentation pH and temperature. Degradation of gliadins catalysed by Lactobacillus acidophilus 5e2 peptidases and a commercial prolyl endopeptidase synthesised by A. niger, carried out at pH 4.0 and 37 ◦C, reduces the gliadin concentration over 110-fold and decreases the relative immunoreactivity of the hydrolysate to 0.9% of its initial value. Hydrolysis of coeliac-toxic peptides: LGQQQPFPPQQPY (P1) and PQPQLPYPQPQLP (P2) under the same conditions occurs with the highest efficiency, reaching 99.8 0.0% and 97.5 0.1%, respectively. ± ± The relative immunoreactivity of peptides P1 and P2 was 0.8 0.0% and 3.2 0.0%, respectively. ± ± A mixture of peptidases from L. acidophilus 5e2 and A. niger may be used in wheat sourdough fermentation to reduce the time needed for degradation of proteins and products of their hydrolysis. -

CDH12 Cadherin 12, Type 2 N-Cadherin 2 RPL5 Ribosomal
5 6 6 5 . 4 2 1 1 1 2 4 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 2 2 A A A A A A A A A A A A A A A A A A A A C C C C C C C C C C C C C C C C C C C C R R R R R R R R R R R R R R R R R R R R B , B B B B B B B B B B B B B B B B B B B , 9 , , , , 4 , , 3 0 , , , , , , , , 6 2 , , 5 , 0 8 6 4 , 7 5 7 0 2 8 9 1 3 3 3 1 1 7 5 0 4 1 4 0 7 1 0 2 0 6 7 8 0 2 5 7 8 0 3 8 5 4 9 0 1 0 8 8 3 5 6 7 4 7 9 5 2 1 1 8 2 2 1 7 9 6 2 1 7 1 1 0 4 5 3 5 8 9 1 0 0 4 2 5 0 8 1 4 1 6 9 0 0 6 3 6 9 1 0 9 0 3 8 1 3 5 6 3 6 0 4 2 6 1 0 1 2 1 9 9 7 9 5 7 1 5 8 9 8 8 2 1 9 9 1 1 1 9 6 9 8 9 7 8 4 5 8 8 6 4 8 1 1 2 8 6 2 7 9 8 3 5 4 3 2 1 7 9 5 3 1 3 2 1 2 9 5 1 1 1 1 1 1 5 9 5 3 2 6 3 4 1 3 1 1 4 1 4 1 7 1 3 4 3 2 7 6 4 2 7 2 1 2 1 5 1 6 3 5 6 1 3 6 4 7 1 6 5 1 1 4 1 6 1 7 6 4 7 e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m -
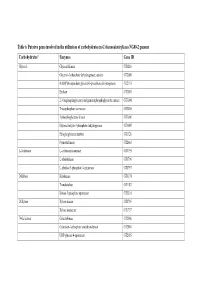
Table 6. Putative Genes Involved in the Utilization of Carbohydrates in G
Table 6. Putative genes involved in the utilization of carbohydrates in G. thermodenitrificans NG80-2 genome Carbohydrates* Enzymes Gene ID Glycerol Glycerol Kinase GT1216 Glycerol-3-phosphate dehydrogenase, aerobic GT2089 NAD(P)H-dependent glycerol-3-phosphate dehydrogenase GT2153 Enolase GT3003 2,3-bisphosphoglycerate-independentphosphoglycerate mutase GT3004 Triosephosphate isomerase GT3005 3-phosphoglycerate kinase GT3006 Glyceraldehyde-3-phosphate dehydrogenase GT3007 Phosphoglycerate mutase GT1326 Pyruvate kinase GT2663 L-Arabinose L-arabinose isomerase GT1795 L-ribulokinase GT1796 L-ribulose 5-phosphate 4-epimerase GT1797 D-Ribose Ribokinase GT3174 Transketolase GT1187 Ribose 5-phosphate epimerase GT3316 D-Xylose Xylose kinase GT1756 Xylose isomerase GT1757 D-Galactose Galactokinase GT2086 Galactose-1-phosphate uridyltransferase GT2084 UDP-glucose 4-epimerase GT2085 Carbohydrates* Enzymes Gene ID D-Fructose 1-phosphofructokinase GT1727 Fructose-1,6-bisphosphate aldolase GT1805 Fructose-1,6-bisphosphate aldolase type II GT3331 Triosephosphate isomerase GT3005 D-Mannose Mannnose-6 phospate isomelase GT3398 6-phospho-1-fructokinase GT2664 D-Mannitol Mannitol-1-phosphate dehydrogenase GT1844 N-Acetylglucosamine N-acetylglucosamine-6-phosphate deacetylase GT2205 N-acetylglucosamine-6-phosphate isomerase GT2204 D-Maltose Alpha-1,4-glucosidase GT0528, GT1643 Sucrose Sucrose phosphorylase GT3215 D-Trehalose Alpha-glucosidase GT1643 Glucose kinase GT2381 Inositol Myo-inositol catabolism protein iolC;5-dehydro-2- GT1807 deoxygluconokinase -

An Investigation on Catalysis of Acylaminoacyl Peptidases
An investigation on catalysis of acylaminoacyl peptidases PhD Thesis András László Kiss Doctorate School of Biology School leader: Prof. Anna Erdei Structural Biochemistry Programme Programme leader: Prof. László Gráf Supervisor: Prof. László Polgár Institute of Enzymology, Biological Research Center, Hungarian Academy of Sciences Budapest 2007 1. Introduction Serine peptidases contain two residues at the active site in addition to the catalytic triad (His, Asp, Ser), which form a cavity called “oxyanion hole” that accommodates the negatively charged oxyanion in the transition state of the catalysis and donate two H- bonds. Enzymes of the prolyl oligopeptidase (POP) family (acylaminoacyl peptidase, prolyl oligopeptidase, dipeptidyl peptidase IV and oligopeptidase B) are extensively studied. These enzymes are larger than classical serine peptidases and are composed of a peptidase domain with an α/β hydrolase fold and a β-propeller domain. POP and oligopeptidase B are monomeric enzymes and endopeptidases, while the exopeptidase acylaminoacyl peptidase and dipeptidyl peptidase IV are tetrameric and dimeric enzymes, respectively. Acylaminoacyl peptidase (AAP) cleaves acylated amino acids from the N-terminus of the N-acylated peptides that plays important role in many biological and disease processes. Human AAP is encoded by the DNF15S2 locus on the short arm of chromosome 3 at the region 21, which suffers deletions in small cell lung carcinomas, and renal carcinomas, resulting in deficiency in the expression of the enzyme. Acylaminoacyl peptidase is also supposed to be involved in the degradation of oxidatively damaged proteins in cells and can be associated with various diseases where damaged proteins aggregate. Its involvement in cataract formation and in the breakdown of immunogenic formylmethionyl-peptides in the digestive tract after bacterial attack was also suggested. -

High-Resolution Mass Spectrometry-Based Approaches for the Detection and Quantification of Peptidase Activity in Plasma
molecules Article High-Resolution Mass Spectrometry-Based Approaches for the Detection and Quantification of Peptidase Activity in Plasma Elisa Maffioli 1,2 , Zhenze Jiang 3, Simona Nonnis 1,2 , Armando Negri 1,2, Valentina Romeo 1, Christopher B. Lietz 3, Vivian Hook 3,4, Giuseppe Ristagno 5, Giuseppe Baselli 6, Erik B. Kistler 7,8 , Federico Aletti 9, Anthony J. O’Donoghue 3,* and Gabriella Tedeschi 1,2,* 1 Department of Veterinary Medicine, University of Milano, 20133 Milano, Italy; elisa.maffi[email protected] (E.M.); [email protected] (S.N.); [email protected] (A.N.); [email protected] (V.R.) 2 Centre for Nanostructured Materials and Interfaces (CIMAINA), University of Milano, 20133 Milano, Italy 3 Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California San Diego, La Jolla, CA 92093, USA; [email protected] (Z.J.); [email protected] (C.B.L.); [email protected] (V.H.) 4 Department of Neurosciences, School of Medicine, University of California San Diego, La Jolla, CA 92093, USA 5 Department of Pathophysiology and Transplantation, University of Milan, 20133 Milan, Italy; [email protected] 6 Dipartimento di Elettronica, Informazione e Bioingegneria, Politecnico di Milano, 20133 Milan, Italy; [email protected] 7 Department of Anesthesiology & Critical Care, University of California San Diego, La Jolla, CA 92093, USA; [email protected] 8 Department of Anesthesiology & Critical Care, VA San Diego HealthCare System, San Diego, CA 92161, USA 9 Department of Bioengineering, University of California San Diego, La Jolla, CA 92093, USA; [email protected] * Correspondence: [email protected] (A.J.O.); [email protected] (G.T.); Tel.: +1-8585345360 (A.J.O.); +39-02-50318127 (G.T.) Academic Editor: Paolo Iadarola Received: 28 July 2020; Accepted: 4 September 2020; Published: 6 September 2020 Abstract: Proteomic technologies have identified 234 peptidases in plasma but little quantitative information about the proteolytic activity has been uncovered. -

Supplementary Table 1: List of the 316 Genes Regulated During Hyperglycemic Euinsulinemic Clamp in Skeletal Muscle
Supplementary Table 1: List of the 316 genes regulated during hyperglycemic euinsulinemic clamp in skeletal muscle. UGCluster Name Symbol Fold Change Cytoband Response to stress Hs.517581 Heme oxygenase (decycling) 1 HMOX1 3.80 22q12 Hs.374950 Metallothionein 1X MT1X 2.20 16q13 Hs.460867 Metallothionein 1B (functional) MT1B 1.70 16q13 Hs.148778 Oxidation resistance 1 OXR1 1.60 8q23 Hs.513626 Metallothionein 1F (functional) MT1F 1.47 16q13 Hs.534330 Metallothionein 2A MT2A 1.45 16q13 Hs.438462 Metallothionein 1H MT1H 1.42 16q13 Hs.523836 Glutathione S-transferase pi GSTP1 -1.74 11q13 Hs.459952 Stannin SNN -1.92 16p13 Immune response, cytokines & related Hs.478275 TNF (ligand) superfamily, member 10 (TRAIL) TNFSF10 1.58 3q26 Hs.278573 CD59 antigen p18-20 (protectin) CD59 1.49 11p13 Hs.534847 Complement component 4B, telomeric C4A 1.47 6p21.3 Hs.535668 Immunoglobulin lambda variable 6-57 IGLV6-57 1.40 22q11.2 Hs.529846 Calcium modulating ligand CAMLG -1.40 5q23 Hs.193516 B-cell CLL/lymphoma 10 BCL10 -1.40 1p22 Hs.840 Indoleamine-pyrrole 2,3 dioxygenase INDO -1.40 8p12-p11 Hs.201083 Mal, T-cell differentiation protein 2 MAL2 -1.44 Hs.522805 CD99 antigen-like 2 CD99L2 -1.45 Xq28 Hs.50002 Chemokine (C-C motif) ligand 19 CCL19 -1.45 9p13 Hs.350268 Interferon regulatory factor 2 binding protein 2 IRF2BP2 -1.47 1q42.3 Hs.567249 Contactin 1 CNTN1 -1.47 12q11-q12 Hs.132807 MHC class I mRNA fragment 3.8-1 3.8-1 -1.48 6p21.3 Hs.416925 Carcinoembryonic antigen-related cell adhesion molecule 19 CEACAM19 -1.49 19q13.31 Hs.89546 Selectin E (endothelial -

Prolyl Endopeptidase from Aspergillus Niger Expressing A
Annex 3 FORM ON WHICH INFORMATION ON THE COMPOUND TO BE EVALUATED BY JECFA IS PROVIDED In completing this form, only brief information is required. The form may be retyped if more space is needed under any one heading provided that the general format is maintained. Name of Compound(s): Acid prolyl endopeptidase from a genetically modified strain of Aspergillus niger Question(s) to be answered by JECFA Safety evaluation when used as processing aid. (kindly provide a brief justification of the request in case of re-evaluations) 1. Proposal for inclusion submitted by: Ministry of Health, Welfare and Sport Nutrition, Health Protection and Prevention Department Parnassusplein 5 2511 VX The Hague P.O. box 20350 2500 EJ The Hague The Netherlands Tel: +31 703407132 2. Name of compound; trade name(s); chemical name(s): Name of compound : Acid prolyl endopeptidase from a genetically modified strain of Aspergillus niger Trade names : BREWERS CLAREX®, MAXIPRO PSP Chemical name : Acid prolyl endopeptidase (EC 3.4.21.xx1) 3. Names and addresses of basic producers: DSM Food Specialties 15 Rue des Comtesses PO Box 239 59472 Seclin Cédex France Tel: 33 320964545 Fax: 33 320964500 4. Has the manufacturer made a commitment to provide data? Yes. 5. Identification of the manufacturer that will be providing data (Please indicate contact person): Dr Jack Reuvers 1 The name proline endopeptidase is also a synonym of prolyl oligopeptidase, EC (IUBMB) number 3.4.21.26. However, in contrast to oligopeptidases, the enzyme also acts on proteins (Edens et al., 2005; Kubota et al., 2005; Takahashi, 2013). -

And Exopeptidases in a Processing Enzyme System: Activation
Proc. Nail. Acad. Sci. USA Vol. 85, pp. 5468-5472, August 1988 Biochemistry Relationship between endo- and exopeptidases in a processing enzyme system: Activation of an endoprotease by the aminopeptidase B-like activity in somatostatin-28 convertase (brain cortex/basic amino acid pairs/peptide substrates/protease inhibitors/prohormone maturation) SOPHIE GOMEZ, PABLO GLUSCHANKOF, AGNES LEPAGE, AND PAUL COHEN Groupe de Neurobiochimie Cellulaire et Moldculaire, Universitd Pierre et Marie Curie, Unitd Associde 554 au Centre National de la Recherche Scientifique, 96 boulevard Raspail, 75006 Paris, France Communicated by I. Robert Lehman, April 8, 1988 (receivedfor review December 15, 1987) ABSTRACT The somatostatin-28 convertase activity in- somatostatin-14 and the amino-terminal dodecapeptide so- volved in vitro in the processing of somatostatin-28 into the matostatin-28-(1-12) (16). neuropeptides somatostatin-28-(1-12) and somatostatin-14 is We have described an endoprotease that cleaves the composed of an endoprotease and a basic aminopeptidase. We peptide bond on the amino side ofthe Arg-Lys doublet in the report herein on the purification to apparent homogeneity of somatostatin-28 sequence (17, 18), releasing the somato- these two constituents and on their functional interrelationship. statin-28-(1-12) fragment and [Arg-2,Lys-1]somatostatin-14. In particular we observed that after various physicochemical The released [Arg-2,Lys-']somatostatin-14 is further proc- treatments, the 90-kDa endoprotease activity was recovered essed by an aminopeptidase B-like activity (18, 19) that is also both at this molecular mass and as a 45-kDa entity. Moreover, present in the preparation. We report herein the purification the production of [Arg2,LysJllsomatostatin-14 from somato- to apparent homogeneity ofthese two activities.