The Pennsylvania State University
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Auxiliary Iron–Sulfur Cofactors in Radical SAM Enzymes☆
Biochimica et Biophysica Acta 1853 (2015) 1316–1334 Contents lists available at ScienceDirect Biochimica et Biophysica Acta journal homepage: www.elsevier.com/locate/bbamcr Review Auxiliary iron–sulfur cofactors in radical SAM enzymes☆ Nicholas D. Lanz a, Squire J. Booker a,b,⁎ a Department of Biochemistry and Molecular Biology, The Pennsylvania State University, University Park, PA 16802, United States b Department of Chemistry, The Pennsylvania State University, University Park, PA 16802, United States article info abstract Article history: A vast number of enzymes are now known to belong to a superfamily known as radical SAM, which all contain a Received 19 September 2014 [4Fe–4S] cluster ligated by three cysteine residues. The remaining, unligated, iron ion of the cluster binds in Received in revised form 15 December 2014 contact with the α-amino and α-carboxylate groups of S-adenosyl-L-methionine (SAM). This binding mode Accepted 6 January 2015 facilitates inner-sphere electron transfer from the reduced form of the cluster into the sulfur atom of SAM, Available online 15 January 2015 resulting in a reductive cleavage of SAM to methionine and a 5′-deoxyadenosyl radical. The 5′-deoxyadenosyl Keywords: radical then abstracts a target substrate hydrogen atom, initiating a wide variety of radical-based transforma- – Radical SAM tions. A subset of radical SAM enzymes contains one or more additional iron sulfur clusters that are required Iron–sulfur cluster for the reactions they catalyze. However, outside of a subset of sulfur insertion reactions, very little is known S-adenosylmethionine about the roles of these additional clusters. This review will highlight the most recent advances in the identifica- Cofactor maturation tion and characterization of radical SAM enzymes that harbor auxiliary iron–sulfur clusters. -

Changes in Soil Microbial Communities After Long-Term Warming Exposure" (2019)
University of Massachusetts Amherst ScholarWorks@UMass Amherst Doctoral Dissertations Dissertations and Theses October 2019 CHANGES IN SOIL MICROBIAL COMMUNITIES AFTER LONG- TERM WARMING EXPOSURE William G. Rodríguez-Reillo University of Massachusetts Amherst Follow this and additional works at: https://scholarworks.umass.edu/dissertations_2 Part of the Environmental Microbiology and Microbial Ecology Commons Recommended Citation Rodríguez-Reillo, William G., "CHANGES IN SOIL MICROBIAL COMMUNITIES AFTER LONG-TERM WARMING EXPOSURE" (2019). Doctoral Dissertations. 1757. https://doi.org/10.7275/15007293 https://scholarworks.umass.edu/dissertations_2/1757 This Open Access Dissertation is brought to you for free and open access by the Dissertations and Theses at ScholarWorks@UMass Amherst. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of ScholarWorks@UMass Amherst. For more information, please contact [email protected]. CHANGES IN SOIL MICROBIAL COMMUNITIES AFTER LONG-TERM WARMING EXPOSURE A Dissertation Presented by WILLIAM GABRIEL RODRÍGUEZ-REILLO Submitted to the Graduate School of the University of Massachusetts Amherst in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY SEPTEMBER 2019 Organismic and Evolutionary Biology © Copyright by William Gabriel Rodríguez-Reillo 2019 All Rights Reserved CHANGES IN SOIL MICROBIAL COMMUNITIES AFTER LONG-TERM WARMING EXPOSURE A Dissertation Presented by WILLIAM GABRIEL RODRÍGUEZ-REILLO Approved as to style and content by: _________________________________________ Jeffrey L. Blanchard, Chair _________________________________________ Courtney Babbitt, Member _________________________________________ David Sela, Member _________________________________________ Kristina Stinson, Member ______________________________________ Paige Warren, Graduate Program Director Organismic and Evolutionary Biology DEDICATION To my parents, William Rodriguez Arce and Carmen L. Reillo Batista. A quienes aún en la distancia me mantuvieron en sus oraciones. -

Metabolic Functions of the Human Gut Microbiota: the Role of Metalloenzymes Cite This: DOI: 10.1039/C8np00074c Lauren J
Natural Product Reports View Article Online REVIEW View Journal Metabolic functions of the human gut microbiota: the role of metalloenzymes Cite this: DOI: 10.1039/c8np00074c Lauren J. Rajakovich and Emily P. Balskus * Covering: up to the end of 2017 The human body is composed of an equal number of human and microbial cells. While the microbial community inhabiting the human gastrointestinal tract plays an essential role in host health, these organisms have also been connected to various diseases. Yet, the gut microbial functions that modulate host biology are not well established. In this review, we describe metabolic functions of the human gut microbiota that involve metalloenzymes. These activities enable gut microbial colonization, mediate interactions with the host, and impact human health and disease. We highlight cases in which enzyme characterization has advanced our understanding of the gut microbiota and examples that illustrate the diverse ways in which Received 14th August 2018 Creative Commons Attribution 3.0 Unported Licence. metalloenzymes facilitate both essential and unique functions of this community. Finally, we analyze Human DOI: 10.1039/c8np00074c Microbiome Project sequencing datasets to assess the distribution of a prominent family of metalloenzymes rsc.li/npr in human-associated microbial communities, guiding future enzyme characterization efforts. 1 Introduction our gut from infancy and plays a critical role in the development 2 Commensal colonization of the human gut: tness and and maintenance of healthy human physiology. It aids in adaptation developing the innate and adaptive immune systems,1,2 2.1 Glycan sulfation provides nutrients and vitamins,3 and protects against path- 4 This article is licensed under a 2.2 Glycan fucosylation ogen invasion. -

2018 Summer Symposium Program
Ofce of UNDERGRADUATE RESEARCH ® THE UNIVERSITY OF UTAH 2018 Summer Symposium THURSDAY, AUGUST 2, 2018 9:00AM - 12:00PM CROCKER SCIENCE CENTER UNIVERSITY OF UTAH 2018 SUMMER SYMPOSIUM Thursday, August 2, 2018 9:00 AM – 12:00 PM Crocker Science Center University of Utah The Office of Undergraduate Research is grateful for the generous support of the Office of the Vice President for Research. We are also thankful for the development of the Summer Research Program Partnership, which is a new collaboration among the Chemistry Research Experience for Undergraduates (REU), the Huntsman Cancer Institute’s PathMaker Cancer Research Program, the Native American Summer Research Internship (NARI), the Physics & Astronomy REU and Summer Undergraduate Research Program, and the Summer Program for Undergraduate Research (SPUR). Together, these programs are serving more than 90 undergraduate researchers in Summer 2018. Finally, we would like to express our utmost pride and congratulations to the students, graduate students, and faculty mentors without whose efforts and dedication this event would not be possible. PROGRAM SCHEDULE NOTE: All student presenters MUST check-in Snacks available at 10:15 AM in Room 205/206 8:30 – 9:00 AM CHECK-IN & POSTER SET-UP 9:00 – 10:30 AM POSTER SESSION I (ODD POSTERS) 10:30 AM – 12:00 PM POSTER SESSION II (EVEN POSTERS) 12:00 – 12:15 PM POSTER TAKE-DOWN 2 SCHEDULE OF PRESENTATIONS POSTER SESSION I 9:00 – 10:30 AM Poster 1 Presenter: Sara Alektiar (University of Michigan) Mentor: Matthew Sigman (Chemistry) Electrocatalytic Bis(bipyidine)ruthenium Hydroxylation of Tertiary and Benzylic C-H Bonds The Sigman and Du Bois labs recently reported a methodology that employs a bis(bipyridine)Ru catalyst operating in acidic water to achieve oxidation of tertiary and benzylic C-H bonds in the presence of basic amines. -
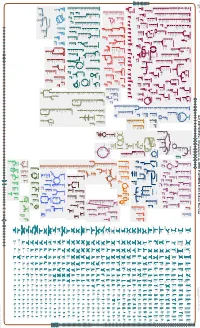
Generated by SRI International Pathway Tools Version 25.0 on Mon
Authors: Pallavi Subhraveti Ron Caspi Peter Midford Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_000716605Cyc: Streptomyces sp. NRRL F-525 NRRL F-525 Cellular Overview Connections between pathways are omitted for legibility. Anamika Kothari an amino an amino an amino an amino an amino an amino an amino an amino phosphate phosphate acid acid acid acid acid acid acid acid molybdate an amino an amino H + a dipeptide phosphate phosphate phosphate acid an amino acid an amino H + phosphate phosphate glycine betaine glycine betaine glycine betaine leu pro pro pro acid acid predicted predicted predicted predicted predicted predicted predicted predicted predicted predicted predicted predicted ABC ABC ABC ABC ABC ABC ABC ABC ABC ABC ABC F0F1 ATP ABC RS16420 RS10685 RS06340 RS46495 RS16425 ProP RS28920 RS26410 LeuE RS27350 RS38790 RS13800 transporter transporter transporter RS41005 transporter transporter transporter transporter transporter CoxB transporter transporter transporter synthase transporter of an of an of an of an of an of an of an of an of molybdate of phosphate of phosphate of a dipeptide amino acid -

Notice: Restrictions
NOTICE: The copyright law of the United States (Title 17, United States Code) governs the making of reproductions of copyrighted material. One specified condition is that the reproduction is not to be "used for any purpose other than private study, scholarship, or research." If a user makes a request for, or later uses a reproduction for purposes in excess of "fair use," that user may be liable for copyright infringement. RESTRICTIONS: This student work may be read, quoted from, cited, for purposes of research. It may not be published in full except by permission of the author. Abstract: The phylogenetic position of a number of bacteria within the family Flavobacteriaceae has been questioned. To address the question, the whole genomes of several organisms were sequenced, and this project is focused on Chryseobacterium haifense. The advances in next generation sequencing (NGS) technologies have caused a decrease in cost for whole genome sequencing. This decreased cost has led to more genomes being sequenced and in the process has caused a large demand for bioinformatics tools to handle the genomic data. To analyze the genomic data, the 930,000 reads were assembled in several steps, using several different software packages to refine the assembly to fewer than 700 contiguous sequences. Automated annotation using the Rapid Annotation using Subsystem Technologies (RAST) server identified the organism’s genes and known pathways which were compared to its phenotypes. The Reciprocal Orthology Score Average (ROSA) genomic similarity calculator showed that Chryseobacterium haifense is as different from “true” Chryseobacteria as other separate genera are which has led to the conclusion that Chryseobacterium haifense does not belong within the Chryseobacterium genus. -

Mechanistic Insights Into the Radical S-Adenosyl L-Methionine Enzyme MFTC
University of Denver Digital Commons @ DU Electronic Theses and Dissertations Graduate Studies 1-1-2017 Mechanistic Insights into the Radical S-Adenosyl L-Methionine Enzyme MFTC Bulat Khaliullin University of Denver Follow this and additional works at: https://digitalcommons.du.edu/etd Part of the Biochemistry, Biophysics, and Structural Biology Commons, and the Chemistry Commons Recommended Citation Khaliullin, Bulat, "Mechanistic Insights into the Radical S-Adenosyl L-Methionine Enzyme MFTC" (2017). Electronic Theses and Dissertations. 1300. https://digitalcommons.du.edu/etd/1300 This Thesis is brought to you for free and open access by the Graduate Studies at Digital Commons @ DU. It has been accepted for inclusion in Electronic Theses and Dissertations by an authorized administrator of Digital Commons @ DU. For more information, please contact [email protected],[email protected]. MECHANISTIC INSIGHTS INTO THE RADICAL S-ADENOSYL L-METHIONINE ENZYME MFTC __________ A Thesis Presented to the Faculty of Natural Sciences and Mathematics University of Denver __________ In Partial Fulfillment of the Requirements for the Degree Master of Science __________ by Bulat Khaliullin June 2017 Advisor: John A. Latham ©Copyright by Bulat Khaliullin 2017 All Rights Reserved Author: Bulat Khaliullin Title: MECHANISTIC INSIGHTS INTO THE RADICAL S-ADENOSYL L- METHIONINE ENZYME MFTC Advisor: John A. Latham Degree Date: June 2017 Abstract Mycofactocin is a putative peptide-derived redox cofactor in Mycobacterium family. Its putative biosynthetic pathway is encoded by the operon mftABCDEF. The initial step of this pathway is a posttranslational modification of a peptide precursor MftA, which is catalyzed by MftC enzyme. This modification only occurs in the presence of chaperone MftB. -

Role of Lysx Gene from Mycobacterium Avium Hominissuis in Metabolism
Role of lysX gene from Mycobacterium avium hominissuis in metabolism and host-cell interaction Inaugural-Dissertation to obtain the academic degree Doctor rerum naturalium (Dr. rer. nat) Submitted to the Department of Biology, Chemistry and Pharmacy of Freie Universität Berlin by GREANA KIRUBAKAR from Chennai, India Berlin, 2019 This work was accomplished between October 2015 and July 2019 at the Robert Koch Institute under the supervision of Dr. Astrid Lewin. 1st Reviewer: Dr. Astrid Lewin 2nd Reviewer: Prof. Dr. Rupert Mutzel Date of Defense: 21.10.2019 Acknowledgement I express my deepest gratitude to my doctoral guide, Dr. Astrid Lewin for giving me the opportunity to work in this prestigious Robert Koch Institute (RKI). Her constant guidance and encouragement has enabled me to successfully pursue this project. I appreciate all her contributions of time, ideas, and funding recommendations to make my PhD. experience productive and stimulating. I would also like to thank Prof. Dr. Lothar Wieler, the President of the Robert Koch-Institute, for providing me permission to carry out my PhD research work. My sincere thanks to my second supervisor, Prof. Dr. Rupert Mutzel, for his insightful comments and valuable suggestions throughout the period of my thesis. I am also very thankful to Elisabeth Kamal, for her excellent technical assistance and taking care of both my experimental and personal issues. I also acknowledge Dr. Toni Aebischer for his valuable advice in my research project and his hard questions which incented me to widen my research from various perspectives. In regards to the Proteomics experiments, I like to thank Dr. -

ABSTRACT FROCK, ANDREW DAVID. Functional Genomics
ABSTRACT FROCK, ANDREW DAVID. Functional Genomics Analysis of the Microbial Physiology and Ecology of Hyperthermophilic Thermotoga Species. (Under the direction of Dr. Robert Kelly.) Generation of renewable fuels through biological conversion of inexpensive carbohydrate feedstocks has been the focus of intense research, which has mostly involved metabolic engineering of model mesophilic microorganisms. However, hyperthermophilic (Topt ≥ 80°C) bacteria of the genus Thermotoga exhibit several desirable traits for a potential biofuel-producing host. They ferment a variety of carbohydrates to hydrogen with high yields approaching the theoretical limit for anaerobic metabolism of 4 mol H2/mol glucose. High temperature growth facilitates biomass feedstock degradation, decreases risk of contamination, and eliminates the reactor cooling costs associated with growth of mesophiles. To further understand the glycoside hydrolase and carbohydrate transporter inventory of these bacteria, four closely-related Thermotoga species were compared. T. maritima, T. neapolitana, T. petrophila, and T. sp. strain RQ2 share approximately 75% of their genomes, yet each species exhibited distinguishing features during growth on simple and complex carbohydrates that correlated with their genotypes. T. maritima and T. neapolitana displayed similar monosaccharide utilization profiles, with a strong preference for glucose and xylose. Only T. sp. strain RQ2 has a fructose specific phosphotransferase system (PTS), and it was the only species found to utilize fructose. T. petrophila consumes glucose to a lesser extent and up-regulates a transporter normally used for xylose (XylE1F1K1) to compensate for the absence of the primary glucose transporter in these species (XylE2F2K2). In some cases, the use of mixed cultures has been proposed to combine the complementary strengths of different microbes for the development of improved bioprocesses. -

Supplementary Material
Supplementary material Figure S1. Cluster analysis of the proteome profile based on qualitative data in low and high sugar conditions. Figure S2. Expression pattern of proteins under high and low sugar cultivation of Granulicella sp. WH15 a) All proteins identified in at least two out of three replicates (excluding on/off proteins). b) Only proteins with significant change t-test p=0.01. 2fold change is indicated by a red line. Figure S3. TigrFam roles of the differentially expressed proteins, excluding proteins with unknown function. Figure S4. General overview of up (red) and downregulated (blue) metabolic pathways based on KEGG analysis of proteome. Table S1. growth of strain Granulicella sp. WH15 in culture media supplemented with different carbon sources. Carbon Source Growth Pectin - Glycogen - Glucosamine - Cellulose - D-glucose + D-galactose + D-mannose + D-xylose + L-arabinose + L-rhamnose + D-galacturonic acid - Cellobiose + D-lactose + Sucrose + +=positive growth; -=No growth. Table S2. Total number of transcripts reads per sample in low and high sugar conditions. Sample ID Total Number of Reads Low sugar (1) 15,731,147 Low sugar (2) 12,624,878 Low sugar (3) 11,080,985 High sugar (1) 11,138,128 High sugar (2) 9,322,795 High sugar (3) 10,071,593 Table S3. Differentially up and down regulated transcripts in high sugar treatment. ORF Annotation Log2FC GWH15_14040 hypothetical protein 3.71 GWH15_06005 hypothetical protein 3.12 GWH15_00285 tRNA-Asn(gtt) 2.74 GWH15_06010 hypothetical protein 2.70 GWH15_14055 hypothetical protein 2.66 -

Generated by SRI International Pathway Tools Version 25.0 on Wed
Authors: Pallavi Subhraveti Peter D Karp Ingrid Keseler An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Anamika Kothari Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_000308495Cyc: Nocardia brevicatena NBRC 12119 Cellular Overview Connections between pathways are omitted for legibility. -

Genome Properties Documentation Release 0.1B
Genome Properties Documentation Release 0.1b Lorna Richardson, Neil Rawlings, Alex Mitchell, Gustavo Salazar Orejuela, Robert D. Finn Jul 10, 2019 Contents 1 About Genome Properties 3 1.1 How to access Genome Properties data.................................3 1.2 Background................................................4 2 PATHWAYS 5 3 METAPATH 13 4 SYSTEM 15 5 COMPLEX 19 6 GUILD 21 7 CATEGORY 23 8 Genome Property Types 25 9 Flatfile Format 27 9.1 DESC file................................................. 27 9.2 FASTA file................................................ 30 9.3 Status file................................................. 31 10 Calculating Genome Properties 33 10.1 Website/Viewer method......................................... 33 10.2 Local analysis method.......................................... 33 11 Contributing to Genome Properties 35 12 Funding 37 i ii Genome Properties Documentation, Release 0.1b Contents: Contents 1 Genome Properties Documentation, Release 0.1b 2 Contents CHAPTER 1 About Genome Properties Genome properties (GP) is an annotation system whereby functional attributes can be assigned to a genome, based on the presence of a defined set of protein family markers within that genome. For example, a species can be proposed to synthesise proline if it can be shown that the genome for that species encodes all the necessary proteins required to carry out the various biochemical steps in the proline biosynthesis pathway. While it is possible to infer this kind of information through analysis of genomic sequence, using protein family models, like those utilised by InterPro, reduces the number of calculations while at the same time increasing the sensitivity of the query. Integrating the GP annotations into InterPro makes the process of calculating a genome property for any given genome/proteome faster and more accurate than by sequence comparison.