ABSTRACT FROCK, ANDREW DAVID. Functional Genomics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Auxiliary Iron–Sulfur Cofactors in Radical SAM Enzymes☆
Biochimica et Biophysica Acta 1853 (2015) 1316–1334 Contents lists available at ScienceDirect Biochimica et Biophysica Acta journal homepage: www.elsevier.com/locate/bbamcr Review Auxiliary iron–sulfur cofactors in radical SAM enzymes☆ Nicholas D. Lanz a, Squire J. Booker a,b,⁎ a Department of Biochemistry and Molecular Biology, The Pennsylvania State University, University Park, PA 16802, United States b Department of Chemistry, The Pennsylvania State University, University Park, PA 16802, United States article info abstract Article history: A vast number of enzymes are now known to belong to a superfamily known as radical SAM, which all contain a Received 19 September 2014 [4Fe–4S] cluster ligated by three cysteine residues. The remaining, unligated, iron ion of the cluster binds in Received in revised form 15 December 2014 contact with the α-amino and α-carboxylate groups of S-adenosyl-L-methionine (SAM). This binding mode Accepted 6 January 2015 facilitates inner-sphere electron transfer from the reduced form of the cluster into the sulfur atom of SAM, Available online 15 January 2015 resulting in a reductive cleavage of SAM to methionine and a 5′-deoxyadenosyl radical. The 5′-deoxyadenosyl Keywords: radical then abstracts a target substrate hydrogen atom, initiating a wide variety of radical-based transforma- – Radical SAM tions. A subset of radical SAM enzymes contains one or more additional iron sulfur clusters that are required Iron–sulfur cluster for the reactions they catalyze. However, outside of a subset of sulfur insertion reactions, very little is known S-adenosylmethionine about the roles of these additional clusters. This review will highlight the most recent advances in the identifica- Cofactor maturation tion and characterization of radical SAM enzymes that harbor auxiliary iron–sulfur clusters. -

Microbial Diversity of Non-Flooded High Temperature Petroleum Reservoir in South of Iran
Archive of SID Biological Journal of Microorganism th 8 Year, Vol. 8, No. 32, Winter 2020 Received: November 18, 2018/ Accepted: May 21, 2019. Page: 15-231- 8 Microbial Diversity of Non-flooded High Temperature Petroleum Reservoir in South of Iran Mohsen Pournia Department of Microbiology, Shiraz Branch, Islamic Azad University, Shiraz, Iran, [email protected] Nima Bahador * Department of Microbiology, Shiraz Branch, Islamic Azad University, Shiraz, Iran, [email protected] Meisam Tabatabaei Biofuel Research Team (BRTeam), Karaj, Iran, [email protected] Reza Azarbayjani Molecular bank, Iranian Biological Resource Center, ACECR, Karaj, Iran, [email protected] Ghassem Hosseni Salekdeh Department of Biology, Agricultural Biotechnology Research Institute, Karaj, Iran, [email protected] Abstract Introduction: Although bacteria and archaea are able to grow and adapted to the petrol reservoirs during several years, there are no results from microbial diversity of oilfields with high temperature in Iran. Hence, the present study tried to identify microbial community in non-water flooding Zeilaei (ZZ) oil reservoir. Materials and methods: In this study, for the first time, non-water flooded high temperature Zeilaei oilfield was analyzed for its microbial community based on next generation sequencing of 16S rRNA genes. Results: The results obtained from this study indicated that the most abundant bacterial community belonged to phylum of Firmicutes (Bacilli ) and Thermotoga, while other phyla (Proteobacteria , Actinobacteria and Synergistetes ) were much less abundant. Bacillus subtilis , B. licheniformis , Petrotoga mobilis , P. miotherma, Fervidobacterium pennivorans , and Thermotoga subterranea were observed with high frequency. In addition, the most abundant archaea were Methanothermobacter thermautotrophicus . Discussion and conclusion: Although there are many reports on the microbial community of oil filed reservoirs, this is the first report of large quantities of Bacillus spp. -

Counts Metabolic Yr10.Pdf
Advanced Review Physiological, metabolic and biotechnological features of extremely thermophilic microorganisms James A. Counts,1 Benjamin M. Zeldes,1 Laura L. Lee,1 Christopher T. Straub,1 Michael W.W. Adams2 and Robert M. Kelly1* The current upper thermal limit for life as we know it is approximately 120C. Microorganisms that grow optimally at temperatures of 75C and above are usu- ally referred to as ‘extreme thermophiles’ and include both bacteria and archaea. For over a century, there has been great scientific curiosity in the basic tenets that support life in thermal biotopes on earth and potentially on other solar bodies. Extreme thermophiles can be aerobes, anaerobes, autotrophs, hetero- trophs, or chemolithotrophs, and are found in diverse environments including shallow marine fissures, deep sea hydrothermal vents, terrestrial hot springs— basically, anywhere there is hot water. Initial efforts to study extreme thermo- philes faced challenges with their isolation from difficult to access locales, pro- blems with their cultivation in laboratories, and lack of molecular tools. Fortunately, because of their relatively small genomes, many extreme thermo- philes were among the first organisms to be sequenced, thereby opening up the application of systems biology-based methods to probe their unique physiologi- cal, metabolic and biotechnological features. The bacterial genera Caldicellulosir- uptor, Thermotoga and Thermus, and the archaea belonging to the orders Thermococcales and Sulfolobales, are among the most studied extreme thermo- philes to date. The recent emergence of genetic tools for many of these organ- isms provides the opportunity to move beyond basic discovery and manipulation to biotechnologically relevant applications of metabolic engineering. -

Changes in Soil Microbial Communities After Long-Term Warming Exposure" (2019)
University of Massachusetts Amherst ScholarWorks@UMass Amherst Doctoral Dissertations Dissertations and Theses October 2019 CHANGES IN SOIL MICROBIAL COMMUNITIES AFTER LONG- TERM WARMING EXPOSURE William G. Rodríguez-Reillo University of Massachusetts Amherst Follow this and additional works at: https://scholarworks.umass.edu/dissertations_2 Part of the Environmental Microbiology and Microbial Ecology Commons Recommended Citation Rodríguez-Reillo, William G., "CHANGES IN SOIL MICROBIAL COMMUNITIES AFTER LONG-TERM WARMING EXPOSURE" (2019). Doctoral Dissertations. 1757. https://doi.org/10.7275/15007293 https://scholarworks.umass.edu/dissertations_2/1757 This Open Access Dissertation is brought to you for free and open access by the Dissertations and Theses at ScholarWorks@UMass Amherst. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of ScholarWorks@UMass Amherst. For more information, please contact [email protected]. CHANGES IN SOIL MICROBIAL COMMUNITIES AFTER LONG-TERM WARMING EXPOSURE A Dissertation Presented by WILLIAM GABRIEL RODRÍGUEZ-REILLO Submitted to the Graduate School of the University of Massachusetts Amherst in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY SEPTEMBER 2019 Organismic and Evolutionary Biology © Copyright by William Gabriel Rodríguez-Reillo 2019 All Rights Reserved CHANGES IN SOIL MICROBIAL COMMUNITIES AFTER LONG-TERM WARMING EXPOSURE A Dissertation Presented by WILLIAM GABRIEL RODRÍGUEZ-REILLO Approved as to style and content by: _________________________________________ Jeffrey L. Blanchard, Chair _________________________________________ Courtney Babbitt, Member _________________________________________ David Sela, Member _________________________________________ Kristina Stinson, Member ______________________________________ Paige Warren, Graduate Program Director Organismic and Evolutionary Biology DEDICATION To my parents, William Rodriguez Arce and Carmen L. Reillo Batista. A quienes aún en la distancia me mantuvieron en sus oraciones. -

Metabolic Functions of the Human Gut Microbiota: the Role of Metalloenzymes Cite This: DOI: 10.1039/C8np00074c Lauren J
Natural Product Reports View Article Online REVIEW View Journal Metabolic functions of the human gut microbiota: the role of metalloenzymes Cite this: DOI: 10.1039/c8np00074c Lauren J. Rajakovich and Emily P. Balskus * Covering: up to the end of 2017 The human body is composed of an equal number of human and microbial cells. While the microbial community inhabiting the human gastrointestinal tract plays an essential role in host health, these organisms have also been connected to various diseases. Yet, the gut microbial functions that modulate host biology are not well established. In this review, we describe metabolic functions of the human gut microbiota that involve metalloenzymes. These activities enable gut microbial colonization, mediate interactions with the host, and impact human health and disease. We highlight cases in which enzyme characterization has advanced our understanding of the gut microbiota and examples that illustrate the diverse ways in which Received 14th August 2018 Creative Commons Attribution 3.0 Unported Licence. metalloenzymes facilitate both essential and unique functions of this community. Finally, we analyze Human DOI: 10.1039/c8np00074c Microbiome Project sequencing datasets to assess the distribution of a prominent family of metalloenzymes rsc.li/npr in human-associated microbial communities, guiding future enzyme characterization efforts. 1 Introduction our gut from infancy and plays a critical role in the development 2 Commensal colonization of the human gut: tness and and maintenance of healthy human physiology. It aids in adaptation developing the innate and adaptive immune systems,1,2 2.1 Glycan sulfation provides nutrients and vitamins,3 and protects against path- 4 This article is licensed under a 2.2 Glycan fucosylation ogen invasion. -

2018 Summer Symposium Program
Ofce of UNDERGRADUATE RESEARCH ® THE UNIVERSITY OF UTAH 2018 Summer Symposium THURSDAY, AUGUST 2, 2018 9:00AM - 12:00PM CROCKER SCIENCE CENTER UNIVERSITY OF UTAH 2018 SUMMER SYMPOSIUM Thursday, August 2, 2018 9:00 AM – 12:00 PM Crocker Science Center University of Utah The Office of Undergraduate Research is grateful for the generous support of the Office of the Vice President for Research. We are also thankful for the development of the Summer Research Program Partnership, which is a new collaboration among the Chemistry Research Experience for Undergraduates (REU), the Huntsman Cancer Institute’s PathMaker Cancer Research Program, the Native American Summer Research Internship (NARI), the Physics & Astronomy REU and Summer Undergraduate Research Program, and the Summer Program for Undergraduate Research (SPUR). Together, these programs are serving more than 90 undergraduate researchers in Summer 2018. Finally, we would like to express our utmost pride and congratulations to the students, graduate students, and faculty mentors without whose efforts and dedication this event would not be possible. PROGRAM SCHEDULE NOTE: All student presenters MUST check-in Snacks available at 10:15 AM in Room 205/206 8:30 – 9:00 AM CHECK-IN & POSTER SET-UP 9:00 – 10:30 AM POSTER SESSION I (ODD POSTERS) 10:30 AM – 12:00 PM POSTER SESSION II (EVEN POSTERS) 12:00 – 12:15 PM POSTER TAKE-DOWN 2 SCHEDULE OF PRESENTATIONS POSTER SESSION I 9:00 – 10:30 AM Poster 1 Presenter: Sara Alektiar (University of Michigan) Mentor: Matthew Sigman (Chemistry) Electrocatalytic Bis(bipyidine)ruthenium Hydroxylation of Tertiary and Benzylic C-H Bonds The Sigman and Du Bois labs recently reported a methodology that employs a bis(bipyridine)Ru catalyst operating in acidic water to achieve oxidation of tertiary and benzylic C-H bonds in the presence of basic amines. -
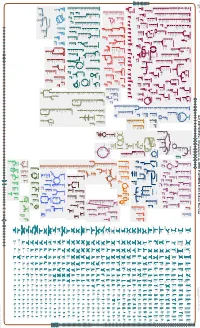
Generated by SRI International Pathway Tools Version 25.0 on Mon
Authors: Pallavi Subhraveti Ron Caspi Peter Midford Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_000716605Cyc: Streptomyces sp. NRRL F-525 NRRL F-525 Cellular Overview Connections between pathways are omitted for legibility. Anamika Kothari an amino an amino an amino an amino an amino an amino an amino an amino phosphate phosphate acid acid acid acid acid acid acid acid molybdate an amino an amino H + a dipeptide phosphate phosphate phosphate acid an amino acid an amino H + phosphate phosphate glycine betaine glycine betaine glycine betaine leu pro pro pro acid acid predicted predicted predicted predicted predicted predicted predicted predicted predicted predicted predicted predicted ABC ABC ABC ABC ABC ABC ABC ABC ABC ABC ABC F0F1 ATP ABC RS16420 RS10685 RS06340 RS46495 RS16425 ProP RS28920 RS26410 LeuE RS27350 RS38790 RS13800 transporter transporter transporter RS41005 transporter transporter transporter transporter transporter CoxB transporter transporter transporter synthase transporter of an of an of an of an of an of an of an of an of molybdate of phosphate of phosphate of a dipeptide amino acid -

Notice: Restrictions
NOTICE: The copyright law of the United States (Title 17, United States Code) governs the making of reproductions of copyrighted material. One specified condition is that the reproduction is not to be "used for any purpose other than private study, scholarship, or research." If a user makes a request for, or later uses a reproduction for purposes in excess of "fair use," that user may be liable for copyright infringement. RESTRICTIONS: This student work may be read, quoted from, cited, for purposes of research. It may not be published in full except by permission of the author. Abstract: The phylogenetic position of a number of bacteria within the family Flavobacteriaceae has been questioned. To address the question, the whole genomes of several organisms were sequenced, and this project is focused on Chryseobacterium haifense. The advances in next generation sequencing (NGS) technologies have caused a decrease in cost for whole genome sequencing. This decreased cost has led to more genomes being sequenced and in the process has caused a large demand for bioinformatics tools to handle the genomic data. To analyze the genomic data, the 930,000 reads were assembled in several steps, using several different software packages to refine the assembly to fewer than 700 contiguous sequences. Automated annotation using the Rapid Annotation using Subsystem Technologies (RAST) server identified the organism’s genes and known pathways which were compared to its phenotypes. The Reciprocal Orthology Score Average (ROSA) genomic similarity calculator showed that Chryseobacterium haifense is as different from “true” Chryseobacteria as other separate genera are which has led to the conclusion that Chryseobacterium haifense does not belong within the Chryseobacterium genus. -

4 Metabolic and Taxonomic Diversification in Continental Magmatic Hydrothermal Systems
Maximiliano J. Amenabar, Matthew R. Urschel, and Eric S. Boyd 4 Metabolic and taxonomic diversification in continental magmatic hydrothermal systems 4.1 Introduction Hydrothermal systems integrate geological processes from the deep crust to the Earth’s surface yielding an extensive array of spring types with an extraordinary diversity of geochemical compositions. Such geochemical diversity selects for unique metabolic properties expressed through novel enzymes and functional characteristics that are tailored to the specific conditions of their local environment. This dynamic interaction between geochemical variation and biology has played out over evolu- tionary time to engender tightly coupled and efficient biogeochemical cycles. The timescales by which these evolutionary events took place, however, are typically in- accessible for direct observation. This inaccessibility impedes experimentation aimed at understanding the causative principles of linked biological and geological change unless alternative approaches are used. A successful approach that is commonly used in geological studies involves comparative analysis of spatial variations to test ideas about temporal changes that occur over inaccessible (i.e. geological) timescales. The same approach can be used to examine the links between biology and environment with the aim of reconstructing the sequence of evolutionary events that resulted in the diversity of organisms that inhabit modern day hydrothermal environments and the mechanisms by which this sequence of events occurred. By combining molecu- lar biological and geochemical analyses with robust phylogenetic frameworks using approaches commonly referred to as phylogenetic ecology [1, 2], it is now possible to take advantage of variation within the present – the distribution of biodiversity and metabolic strategies across geochemical gradients – to recognize the extent of diversity and the reasons that it exists. -

Complex Subsurface Hydrothermal Fluid Mixing at a Submarine Arc Volcano Supports Distinct and Highly Diverse Microbial Communities
Complex subsurface hydrothermal fluid mixing at a submarine arc volcano supports distinct and highly diverse microbial communities Anna-Louise Reysenbacha,1,2, Emily St. Johna,2, Jennifer Meneghina, Gilberto E. Floresb, Mircea Podarc, Nina Dombrowskid, Anja Spangd,e, Stephane L’Haridonf, Susan E. Humphrisg, Cornel E. J. de Rondeh, Fabio Caratori Tontinih, Maurice Tiveyg, Valerie K. Stuckeri, Lucy C. Stewarth,j, Alexander Diehlk,l, and Wolfgang Bachk,l aCenter for Life in Extreme Environments, Biology Department, Portland State University, Portland, OR 97201; bDepartment of Biology, California State University, Northridge, CA 91330; cBiosciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831; dRoyal Netherlands Institute for Sea Research, Department of Marine Microbiology and Biogeochemistry, NL-1790 AB Den Burg, The Netherlands; eDepartment of Cell and Molecular Biology, Science for Life Laboratory, Uppsala University, SE-75123 Uppsala, Sweden; fCNRS, Institut Français de Recherche pour l’Exploitation de la Mer, Laboratoire de Microbiologie des Environnements Extrêmes, Université de Bretagne Occidentale, F-29280 Plouzané, France; gDepartment of Geology and Geophysics, Woods Hole Oceanographic Institution, Woods Hole, MA 02543; hDepartment of Earth Systems and Resources, GNS Science, Avalon, Lower Hutt 5010, New Zealand; iLaboratories and Collections, GNS Science, Avalon, Lower Hutt 5010, New Zealand; jToha Science, Wellington 6011, New Zealand; kFaculty of Geosciences, University of Bremen, 28359 Bremen, Germany; and lMARUM - Center for Marine Environmental Sciences, University of Bremen, 28359 Bremen, Germany Edited by Edward F. DeLong, University of Hawaii at Manoa, Honolulu, HI, and approved November 3, 2020 (received for review September 10, 2020) Hydrothermally active submarine volcanoes are mineral-rich bio- seawater circulation through oceanic crust. -

Bibliography
Bibliography Abella, C.A., X.P. Cristina, A. Martinez, I. Pibernat and X. Vila. 1998. on moderate concentrations of acetate: production of single cells. Two new motile phototrophic consortia: "Chlorochromatium lunatum" Appl. Microbiol. Biotechnol. 35: 686-689. and "Pelochromatium selenoides". Arch. Microbiol. 169: 452-459. Ahring, B.K, P. Westermann and RA. Mah. 1991b. Hydrogen inhibition Abella, C.A and LJ. Garcia-Gil. 1992. Microbial ecology of planktonic of acetate metabolism and kinetics of hydrogen consumption by Me filamentous phototrophic bacteria in holomictic freshwater lakes. Hy thanosarcina thermophila TM-I. Arch. Microbiol. 157: 38-42. drobiologia 243-244: 79-86. Ainsworth, G.C. and P.H.A Sheath. 1962. Microbial Classification: Ap Acca, M., M. Bocchetta, E. Ceccarelli, R Creti, KO. Stetter and P. Cam pendix I. Symp. Soc. Gen. Microbiol. 12: 456-463. marano. 1994. Updating mass and composition of archaeal and bac Alam, M. and D. Oesterhelt. 1984. Morphology, function and isolation terial ribosomes. Archaeal-like features of ribosomes from the deep of halobacterial flagella. ]. Mol. Biol. 176: 459-476. branching bacterium Aquifex pyrophilus. Syst. Appl. Microbiol. 16: 629- Albertano, P. and L. Kovacik. 1994. Is the genus LeptolynglYya (Cyano 637. phyte) a homogeneous taxon? Arch. Hydrobiol. Suppl. 105: 37-51. Achenbach-Richter, L., R Gupta, KO. Stetter and C.R Woese. 1987. Were Aldrich, H.C., D.B. Beimborn and P. Schönheit. 1987. Creation of arti the original eubacteria thermophiles? Syst. Appl. Microbiol. 9: 34- factual internal membranes during fixation of Methanobacterium ther 39. moautotrophicum. Can.]. Microbiol. 33: 844-849. Adams, D.G., D. Ashworth and B. -

Development of Genetic Tools for Thermotoga Spp
DEVELOPMENT OF GENETIC TOOLS FOR THERMOTOGA SPP. Dongmei Han A Dissertation Submitted to the Graduate College of Bowling Green State University in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY December 2013 Committee: Dr. Zhaohui Xu, Advisor Dr. Lisa C. Chavers Graduate Faculty Representative Dr. George S. Bullerjahn Dr. Raymond A. Larsen Dr. Scott O. Rogers © 2013 Dongmei Han All Rights Reserved iii ABSTRACT Zhaohui Xu, Advisor Thermotoga spp. may serve as model systems for understanding life sustainability under hyperthermophilic conditions. They are also attractive candidates for producing biohydrogen in industry. However, a lack of genetic tools has hampered the investigation and application of these organisms. We improved the cultivation method of Thermotoga spp. for preparing and handling Thermotoga solid cultures under aerobic conditions. An embedded method achieved a plating efficiency of ~ 50%, and a soft SVO medium was introduced to bridge isolating single Thermotoga colonies from solid medium to liquid medium. The morphological change of T. neapolitana during the growth process was observed through scanning electron microscopy and transmission electron microscopy. At the early exponential phase, around OD600 0.1 – 0.2, the area of adhered region between toga and cell membrane was the largest, and it was suspected to be the optimal time for DNA uptake in transformation. The capacity of natural transformation was found in T. sp. RQ7, but not in T. maritima. A Thermotoga-E. coli shuttle vector pDH10 was constructed using pRQ7, a cryptic mini-plasmid isolated from T. sp. RQ7. Plasmid pDH10 was introduced to T. sp. RQ7 by liposome-mediated transformation, electroporation, and natural transformation, and to T.