Changes in Soil Microbial Communities After Long-Term Warming Exposure" (2019)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Consistent Responses of Soil Microbial Communities to Elevated Nutrient Inputs in Grasslands Across the Globe
Consistent responses of soil microbial communities to elevated nutrient inputs in grasslands across the globe Jonathan W. Leffa,b, Stuart E. Jonesc, Suzanne M. Proberd, Albert Barberána, Elizabeth T. Borere, Jennifer L. Firnf, W. Stanley Harpoleg,h,i, Sarah E. Hobbiee, Kirsten S. Hofmockelj, Johannes M. H. Knopsk, Rebecca L. McCulleyl, Kimberly La Pierrem, Anita C. Rischn, Eric W. Seabloomo, Martin Schützn, Christopher Steenbockb, Carly J. Stevensp, and Noah Fierera,b,1 aCooperative Institute for Research in Environmental Sciences, University of Colorado, Boulder, CO 80309; bDepartment of Ecology and Evolutionary Biology, University of Colorado, Boulder, CO 80309; cDepartment of Biological Sciences, University of Notre Dame, Notre Dame, IN 46556; dCommonwealth Scientific and Industrial Research Organisation Land and Water Flagship, Wembley, WA 6913, Australia; eDepartment of Ecology, Evolution and Behavior, University of Minnesota, St. Paul, MN 55108; fSchool of Earth, Environmental and Biological Sciences, Queensland University of Technology, Brisbane, QLD 4001, Australia; gDepartment of Physiological Diversity, Helmholtz Center for Environmental Research UFZ, 04318 Leipzig, Germany; hGerman Centre for Integrative Biodiversity Research Halle-Jena-Leipzig, D-04103 Leipzig, Germany; iInstitute of Biology, Martin Luther University Halle-Wittenberg, 06108 Halle (Saale), Germany; jEcology, Evolution, and Organismal Biology Department, Iowa State University, Ames, IA 50011; kSchool of Biological Sciences, University of Nebraska, Lincoln, NE 68588; lDepartment of Plant and Soil Sciences, University of Kentucky, Lexington, KY 40546; mDepartment of Integrative Biology, University of California, Berkeley, CA 94720; nCommunity Ecology, Swiss Federal Institute for Forest, Snow and Landscape Research, 8903 Birmensdorf, Switzerland; oDepartment of Ecology, Evolution, and Behavior, University of Minnesota, St. Paul, MN 55108; and pLancaster Environment Centre, Lancaster University, Lancaster, LA1 4YQ, United Kingdom Edited by Peter M. -

Auxiliary Iron–Sulfur Cofactors in Radical SAM Enzymes☆
Biochimica et Biophysica Acta 1853 (2015) 1316–1334 Contents lists available at ScienceDirect Biochimica et Biophysica Acta journal homepage: www.elsevier.com/locate/bbamcr Review Auxiliary iron–sulfur cofactors in radical SAM enzymes☆ Nicholas D. Lanz a, Squire J. Booker a,b,⁎ a Department of Biochemistry and Molecular Biology, The Pennsylvania State University, University Park, PA 16802, United States b Department of Chemistry, The Pennsylvania State University, University Park, PA 16802, United States article info abstract Article history: A vast number of enzymes are now known to belong to a superfamily known as radical SAM, which all contain a Received 19 September 2014 [4Fe–4S] cluster ligated by three cysteine residues. The remaining, unligated, iron ion of the cluster binds in Received in revised form 15 December 2014 contact with the α-amino and α-carboxylate groups of S-adenosyl-L-methionine (SAM). This binding mode Accepted 6 January 2015 facilitates inner-sphere electron transfer from the reduced form of the cluster into the sulfur atom of SAM, Available online 15 January 2015 resulting in a reductive cleavage of SAM to methionine and a 5′-deoxyadenosyl radical. The 5′-deoxyadenosyl Keywords: radical then abstracts a target substrate hydrogen atom, initiating a wide variety of radical-based transforma- – Radical SAM tions. A subset of radical SAM enzymes contains one or more additional iron sulfur clusters that are required Iron–sulfur cluster for the reactions they catalyze. However, outside of a subset of sulfur insertion reactions, very little is known S-adenosylmethionine about the roles of these additional clusters. This review will highlight the most recent advances in the identifica- Cofactor maturation tion and characterization of radical SAM enzymes that harbor auxiliary iron–sulfur clusters. -

Mycosphere Notes 225–274: Types and Other Specimens of Some Genera of Ascomycota
Mycosphere 9(4): 647–754 (2018) www.mycosphere.org ISSN 2077 7019 Article Doi 10.5943/mycosphere/9/4/3 Copyright © Guizhou Academy of Agricultural Sciences Mycosphere Notes 225–274: types and other specimens of some genera of Ascomycota Doilom M1,2,3, Hyde KD2,3,6, Phookamsak R1,2,3, Dai DQ4,, Tang LZ4,14, Hongsanan S5, Chomnunti P6, Boonmee S6, Dayarathne MC6, Li WJ6, Thambugala KM6, Perera RH 6, Daranagama DA6,13, Norphanphoun C6, Konta S6, Dong W6,7, Ertz D8,9, Phillips AJL10, McKenzie EHC11, Vinit K6,7, Ariyawansa HA12, Jones EBG7, Mortimer PE2, Xu JC2,3, Promputtha I1 1 Department of Biology, Faculty of Science, Chiang Mai University, Chiang Mai 50200, Thailand 2 Key Laboratory for Plant Diversity and Biogeography of East Asia, Kunming Institute of Botany, Chinese Academy of Sciences, 132 Lanhei Road, Kunming 650201, China 3 World Agro Forestry Centre, East and Central Asia, 132 Lanhei Road, Kunming 650201, Yunnan Province, People’s Republic of China 4 Center for Yunnan Plateau Biological Resources Protection and Utilization, College of Biological Resource and Food Engineering, Qujing Normal University, Qujing, Yunnan 655011, China 5 Shenzhen Key Laboratory of Microbial Genetic Engineering, College of Life Sciences and Oceanography, Shenzhen University, Shenzhen 518060, China 6 Center of Excellence in Fungal Research, Mae Fah Luang University, Chiang Rai 57100, Thailand 7 Department of Entomology and Plant Pathology, Faculty of Agriculture, Chiang Mai University, Chiang Mai 50200, Thailand 8 Department Research (BT), Botanic Garden Meise, Nieuwelaan 38, BE-1860 Meise, Belgium 9 Direction Générale de l'Enseignement non obligatoire et de la Recherche scientifique, Fédération Wallonie-Bruxelles, Rue A. -

Schoch CL, Crous PW, Groenewald JZS, Boehm EWA, Burgessti
Schoch CL, Crous PW, Groenewald JZS, Boehm EWA, BurgessTI, Gruyter J De, Hoog GS De, Dixon LJ, Grube M, Gueidan C, Harada Y, Hatakeyama S, Hirayama K, Hosoya T, Huhndorf SM, Hyde KD, Jones EBG, Kohlmeyer J, Kruys Å, Li YM, Lücking R, Lumbsch HT, Marvanová L, Mbatchou JS, McVay AH, Miller AN, Mugambi GK, Muggia L, Nelsen MP, Nelson P, Owensby CA, Phillips AJL, Phongpaichit S, Pointing SB, Pujade-Renaud V, Raja HA, Rivas Plata E, Robbertse B, Ruibal C, Sakayaroj J, Sano T, Selbmann L, Shearer CA, Shirouzu T, Slippers B, Suetrong S, Tanaka K, Volkmann-Kohlmeyer B, Wingfield MJ, Wood AR, Woudenberg JHC, Yonezawa H, Zhang Y, Spatafora JW (2009). A class-wide phylogenetic assessment of Dothideomycetes. Studies in Mycology 64: 1–15. Crous PW, Schoch CL, Hyde KD, Wood AR, Gueidan C, Hoog GS De, Groenewald JZ (2009). Phylogenetic lineages in the Capnodiales. Studies in Mycology 64: 17–47. Boehm EWA, Mugambi GK, Miller AN, Huhndorf SM, Marincowitz S, Spatafora JW, Schoch CL (2009). A molecular phylogenetic reappraisal of the Hysteriaceae, Mytilinidiaceae and Gloniaceae (Pleosporomycetidae, Dothideomycetes) with keys to world species. Studies in Mycology 64: 49–83. Zhang Y, Schoch CL, Fournier J, Crous PW, Gruyter J De, Woudenberg JHC, Hirayama K, Tanaka K, Pointing SB, Hyde KD (2009). Multi-locus phylogeny of the Pleosporales: a taxonomic, ecological and evolutionary re-evaluation. Studies in Mycology 64: 85–102. Mugambi GK, Huhndorf SM (2009). Molecular phylogenetics of Pleosporales: Melanommataceae and Lophiostomataceae re-circumscribed (Pleosporomycetidae, Dothideomycetes, Ascomycota). Studies in Mycology 64: 103–121. Ruibal C, Gueidan C, Selbmann L, Gorbushina AA, Crous PW, Groenewald JZ, Muggia L, Grube M, Isola D, Schoch CL, Staley JT, Lutzoni F, Hoog GS De (2009). -

Ectomycorrhizal Fungal Community Structure in a Young Orchard of Grafted and Ungrafted Hybrid Chestnut Saplings
Mycorrhiza (2021) 31:189–201 https://doi.org/10.1007/s00572-020-01015-0 ORIGINAL ARTICLE Ectomycorrhizal fungal community structure in a young orchard of grafted and ungrafted hybrid chestnut saplings Serena Santolamazza‑Carbone1,2 · Laura Iglesias‑Bernabé1 · Esteban Sinde‑Stompel3 · Pedro Pablo Gallego1,2 Received: 29 August 2020 / Accepted: 17 December 2020 / Published online: 27 January 2021 © The Author(s) 2021 Abstract Ectomycorrhizal (ECM) fungal community of the European chestnut has been poorly investigated, and mostly by sporocarp sampling. We proposed the study of the ECM fungal community of 2-year-old chestnut hybrids Castanea × coudercii (Castanea sativa × Castanea crenata) using molecular approaches. By using the chestnut hybrid clones 111 and 125, we assessed the impact of grafting on ECM colonization rate, species diversity, and fungal community composition. The clone type did not have an impact on the studied variables; however, grafting signifcantly infuenced ECM colonization rate in clone 111. Species diversity and richness did not vary between the experimental groups. Grafted and ungrafted plants of clone 111 had a diferent ECM fungal species composition. Sequence data from ITS regions of rDNA revealed the presence of 9 orders, 15 families, 19 genera, and 27 species of ECM fungi, most of them generalist, early-stage species. Thirteen new taxa were described in association with chestnuts. The basidiomycetes Agaricales (13 taxa) and Boletales (11 taxa) represented 36% and 31%, of the total sampled ECM fungal taxa, respectively. Scleroderma citrinum, S. areolatum, and S. polyrhizum (Boletales) were found in 86% of the trees and represented 39% of total ECM root tips. The ascomycete Cenococcum geophilum (Mytilinidiales) was found in 80% of the trees but accounted only for 6% of the colonized root tips. -

Ectomycorrhizal Ecology Is Imprinted in the Genome of the Dominant Symbiotic Fungus Cenococcum Geophilum Martina Peter, Annegret Kohler, Robin A
Ectomycorrhizal ecology is imprinted in the genome of the dominant symbiotic fungus Cenococcum geophilum Martina Peter, Annegret Kohler, Robin A. Ohm, Alan Kuo, Jennifer Kruetzmann, Emmanuelle Morin, Matthias Arend, Kerrie W. Barry, Manfred Binder, Cindy Choi, et al. To cite this version: Martina Peter, Annegret Kohler, Robin A. Ohm, Alan Kuo, Jennifer Kruetzmann, et al.. Ectomycor- rhizal ecology is imprinted in the genome of the dominant symbiotic fungus Cenococcum geophilum. Nature Communications, Nature Publishing Group, 2016, 7, pp.1-15. 10.1038/ncomms12662. hal- 01439098 HAL Id: hal-01439098 https://hal.archives-ouvertes.fr/hal-01439098 Submitted on 7 Jan 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License ARTICLE Received 4 Nov 2015 | Accepted 21 Jul 2016 | Published 7 Sep 2016 DOI: 10.1038/ncomms12662 OPEN Ectomycorrhizal ecology is imprinted in the genome of the dominant symbiotic fungus Cenococcum geophilum Martina Peter1,*, Annegret Kohler2,*, Robin A. Ohm3,4, Alan Kuo3, Jennifer Kru¨tzmann5, Emmanuelle Morin2, Matthias Arend1, Kerrie W. Barry3, Manfred Binder6, Cindy Choi3, Alicia Clum3, Alex Copeland3, Nadine Grisel1, Sajeet Haridas3, Tabea Kipfer1, Kurt LaButti3, Erika Lindquist3, Anna Lipzen3, Renaud Maire1, Barbara Meier1, Sirma Mihaltcheva3, Virginie Molinier1, Claude Murat2, Stefanie Po¨ggeler7,8, C. -

Metabolic Functions of the Human Gut Microbiota: the Role of Metalloenzymes Cite This: DOI: 10.1039/C8np00074c Lauren J
Natural Product Reports View Article Online REVIEW View Journal Metabolic functions of the human gut microbiota: the role of metalloenzymes Cite this: DOI: 10.1039/c8np00074c Lauren J. Rajakovich and Emily P. Balskus * Covering: up to the end of 2017 The human body is composed of an equal number of human and microbial cells. While the microbial community inhabiting the human gastrointestinal tract plays an essential role in host health, these organisms have also been connected to various diseases. Yet, the gut microbial functions that modulate host biology are not well established. In this review, we describe metabolic functions of the human gut microbiota that involve metalloenzymes. These activities enable gut microbial colonization, mediate interactions with the host, and impact human health and disease. We highlight cases in which enzyme characterization has advanced our understanding of the gut microbiota and examples that illustrate the diverse ways in which Received 14th August 2018 Creative Commons Attribution 3.0 Unported Licence. metalloenzymes facilitate both essential and unique functions of this community. Finally, we analyze Human DOI: 10.1039/c8np00074c Microbiome Project sequencing datasets to assess the distribution of a prominent family of metalloenzymes rsc.li/npr in human-associated microbial communities, guiding future enzyme characterization efforts. 1 Introduction our gut from infancy and plays a critical role in the development 2 Commensal colonization of the human gut: tness and and maintenance of healthy human physiology. It aids in adaptation developing the innate and adaptive immune systems,1,2 2.1 Glycan sulfation provides nutrients and vitamins,3 and protects against path- 4 This article is licensed under a 2.2 Glycan fucosylation ogen invasion. -

2018 Summer Symposium Program
Ofce of UNDERGRADUATE RESEARCH ® THE UNIVERSITY OF UTAH 2018 Summer Symposium THURSDAY, AUGUST 2, 2018 9:00AM - 12:00PM CROCKER SCIENCE CENTER UNIVERSITY OF UTAH 2018 SUMMER SYMPOSIUM Thursday, August 2, 2018 9:00 AM – 12:00 PM Crocker Science Center University of Utah The Office of Undergraduate Research is grateful for the generous support of the Office of the Vice President for Research. We are also thankful for the development of the Summer Research Program Partnership, which is a new collaboration among the Chemistry Research Experience for Undergraduates (REU), the Huntsman Cancer Institute’s PathMaker Cancer Research Program, the Native American Summer Research Internship (NARI), the Physics & Astronomy REU and Summer Undergraduate Research Program, and the Summer Program for Undergraduate Research (SPUR). Together, these programs are serving more than 90 undergraduate researchers in Summer 2018. Finally, we would like to express our utmost pride and congratulations to the students, graduate students, and faculty mentors without whose efforts and dedication this event would not be possible. PROGRAM SCHEDULE NOTE: All student presenters MUST check-in Snacks available at 10:15 AM in Room 205/206 8:30 – 9:00 AM CHECK-IN & POSTER SET-UP 9:00 – 10:30 AM POSTER SESSION I (ODD POSTERS) 10:30 AM – 12:00 PM POSTER SESSION II (EVEN POSTERS) 12:00 – 12:15 PM POSTER TAKE-DOWN 2 SCHEDULE OF PRESENTATIONS POSTER SESSION I 9:00 – 10:30 AM Poster 1 Presenter: Sara Alektiar (University of Michigan) Mentor: Matthew Sigman (Chemistry) Electrocatalytic Bis(bipyidine)ruthenium Hydroxylation of Tertiary and Benzylic C-H Bonds The Sigman and Du Bois labs recently reported a methodology that employs a bis(bipyridine)Ru catalyst operating in acidic water to achieve oxidation of tertiary and benzylic C-H bonds in the presence of basic amines. -

Implications of Streamlining Theory for Microbial Ecology
The ISME Journal (2014) 8, 1553–1565 & 2014 International Society for Microbial Ecology All rights reserved 1751-7362/14 www.nature.com/ismej WINOGRADSKY REVIEW Implications of streamlining theory for microbial ecology Stephen J Giovannoni1, J Cameron Thrash1,2 and Ben Temperton1,3 1Department of Microbiology, Oregon State University, Corvallis, OR, USA; 2Department of Biological Sciences, Louisiana State University, Baton Rouge, LA, USA and 3Plymouth Marine Laboratory, Prospect Place, Plymouth, UK Whether a small cell, a small genome or a minimal set of chemical reactions with self-replicating properties, simplicity is beguiling. As Leonardo da Vinci reportedly said, ‘simplicity is the ultimate sophistication’. Two diverging views of simplicity have emerged in accounts of symbiotic and commensal bacteria and cosmopolitan free-living bacteria with small genomes. The small genomes of obligate insect endosymbionts have been attributed to genetic drift caused by small effective population sizes (Ne). In contrast, streamlining theory attributes small cells and genomes to selection for efficient use of nutrients in populations where Ne is large and nutrients limit growth. Regardless of the cause of genome reduction, lost coding potential eventually dictates loss of function. Consequences of reductive evolution in streamlined organisms include atypical patterns of prototrophy and the absence of common regulatory systems, which have been linked to difficulty in culturing these cells. Recent evidence from metagenomics suggests that streamlining is commonplace, may broadly explain the phenomenon of the uncultured microbial majority, and might also explain the highly interdependent (connected) behavior of many microbial ecosystems. Streamlining theory is belied by the observation that many successful bacteria are large cells with complex genomes. -
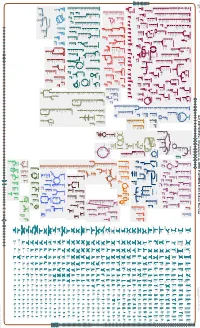
Generated by SRI International Pathway Tools Version 25.0 on Mon
Authors: Pallavi Subhraveti Ron Caspi Peter Midford Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_000716605Cyc: Streptomyces sp. NRRL F-525 NRRL F-525 Cellular Overview Connections between pathways are omitted for legibility. Anamika Kothari an amino an amino an amino an amino an amino an amino an amino an amino phosphate phosphate acid acid acid acid acid acid acid acid molybdate an amino an amino H + a dipeptide phosphate phosphate phosphate acid an amino acid an amino H + phosphate phosphate glycine betaine glycine betaine glycine betaine leu pro pro pro acid acid predicted predicted predicted predicted predicted predicted predicted predicted predicted predicted predicted predicted ABC ABC ABC ABC ABC ABC ABC ABC ABC ABC ABC F0F1 ATP ABC RS16420 RS10685 RS06340 RS46495 RS16425 ProP RS28920 RS26410 LeuE RS27350 RS38790 RS13800 transporter transporter transporter RS41005 transporter transporter transporter transporter transporter CoxB transporter transporter transporter synthase transporter of an of an of an of an of an of an of an of an of molybdate of phosphate of phosphate of a dipeptide amino acid -

Notice: Restrictions
NOTICE: The copyright law of the United States (Title 17, United States Code) governs the making of reproductions of copyrighted material. One specified condition is that the reproduction is not to be "used for any purpose other than private study, scholarship, or research." If a user makes a request for, or later uses a reproduction for purposes in excess of "fair use," that user may be liable for copyright infringement. RESTRICTIONS: This student work may be read, quoted from, cited, for purposes of research. It may not be published in full except by permission of the author. Abstract: The phylogenetic position of a number of bacteria within the family Flavobacteriaceae has been questioned. To address the question, the whole genomes of several organisms were sequenced, and this project is focused on Chryseobacterium haifense. The advances in next generation sequencing (NGS) technologies have caused a decrease in cost for whole genome sequencing. This decreased cost has led to more genomes being sequenced and in the process has caused a large demand for bioinformatics tools to handle the genomic data. To analyze the genomic data, the 930,000 reads were assembled in several steps, using several different software packages to refine the assembly to fewer than 700 contiguous sequences. Automated annotation using the Rapid Annotation using Subsystem Technologies (RAST) server identified the organism’s genes and known pathways which were compared to its phenotypes. The Reciprocal Orthology Score Average (ROSA) genomic similarity calculator showed that Chryseobacterium haifense is as different from “true” Chryseobacteria as other separate genera are which has led to the conclusion that Chryseobacterium haifense does not belong within the Chryseobacterium genus. -

Effects of Various Long-Term Tillage Systems on Some Chemical and Biological Properties of Soil
Pol. J. Environ. Stud. Vol. 22, No. 6 (2013), 1835-1844 Original Research Effects of Various Long-Term Tillage Systems on Some Chemical and Biological Properties of Soil Dorota Swędrzyńska1*, Irena Małecka2, Andrzej Blecharczyk2, Arkadiusz Swędrzyński3, Justyna Starzyk1 1Department of General and Environmental Microbiology, Szydłowska 50, 60-656 Poznań 2Department of Agronomy, Dojazd 11, 60-632 Poznań 3Department of Grassland and Natural Landscape Sciences, Dojazd 11, 60-632 Poznań Poznań University of Life Sciences, Poland Received: 7 July 2013 Accepted: 30 October 2013 Abstract One of the directions of actions limiting the occurrence in soil environments of unfavorable phenome- na accompanying conventional tillage is the introduction of reduced tillage, even total abandonment of culti- vation operations. The objective of the performed investigation was to compare the impact of conventional tillage, reduced tillage, and no-tillage on some soil chemical (Corg., total N, pH) and microbiological (total bac- terial counts, numbers of oligotrophs, copiotrophs and fungi, activity of dehydrogenases and acid phosphatase) properties. Studies carried out in 2010-12 involved a static field experiment initiated in 1999 at Brody Research Station of Poznań University of Life Science, Poland, in temperate climate, on a soil classified as Albic Luvisols developed on loamy sands overlying loamy material. Analyses were performed on soil samples col- lected from under winter wheat from two horizons: 0-10 cm and 10-20 cm. In the 0-10 cm layer, the lowest values of almost all analyzed indices (Corg., total N, total bacterial counts, numbers of oligotrophs, copiotrophs and fungi, as well as the activity of dehydrogenases and acid phos- phatase) were determined in conditions of conventional tillage.