Reactions of Polyfluorobenzenes with Nucleophilic Reagentsl
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ionic Compound Ratios Time: 1 -2 Class Periods
Collisions Lesson Plan Ionic Compound Ratios Time: 1 -2 class periods Lesson Description In this lesson, students will use Collisions to explore the formation of ionic compounds and compound ratios. Key Essential Questions 1. What makes up an ionic compound? 2. Are ionic compounds found in common ratios? Learning Outcomes Students will be able to determine the ionic compound ratio of an ionic compound. Prior Student Knowledge Expected Cations are postiviely charged ions and anions are negatively charged ions. Lesson Materials • Individual student access to Collisions on tablet, Chromebook, or computer. • Projector / display of teacher screen • Accompanying student resources (attached) Standards Alignment NGSS Alignment Science & Enginnering Practices Disciplinary Core Ideas Crosscutting Concepts • Developing and using • HS-PS-12. Construct and • Structure and Function models revise an explanation for the • Construcing explanations outcome of a simple chemical and designing solutions rection based on the outermost electron states of atoms, trends int he periodic table, and knowl- edge of the partterns of chemi- cal properties. www.playmadagames.com ©2018 PlayMada Games LLC. All rights reserved. 1 PART 1: Explore (15 minutes) Summary This is an inquiry-driven activity where students will complete the first few levels of the Collisions Ionic Bonding game to become introduced to the concept of ionic bonding and compound ratios. Activity 1. Direct students to log into Collisions with their individual username and password. 2. Students should enter the Ionic Bonding game and play Levels 1-6 levels. 3. Have your students answer the following questions during gameplay: 1. What combination of ions did you use to successfully match a target? 2. -

Use of Chlorofluorocarbons in Hydrology : a Guidebook
USE OF CHLOROFLUOROCARBONS IN HYDROLOGY A Guidebook USE OF CHLOROFLUOROCARBONS IN HYDROLOGY A GUIDEBOOK 2005 Edition The following States are Members of the International Atomic Energy Agency: AFGHANISTAN GREECE PANAMA ALBANIA GUATEMALA PARAGUAY ALGERIA HAITI PERU ANGOLA HOLY SEE PHILIPPINES ARGENTINA HONDURAS POLAND ARMENIA HUNGARY PORTUGAL AUSTRALIA ICELAND QATAR AUSTRIA INDIA REPUBLIC OF MOLDOVA AZERBAIJAN INDONESIA ROMANIA BANGLADESH IRAN, ISLAMIC REPUBLIC OF RUSSIAN FEDERATION BELARUS IRAQ SAUDI ARABIA BELGIUM IRELAND SENEGAL BENIN ISRAEL SERBIA AND MONTENEGRO BOLIVIA ITALY SEYCHELLES BOSNIA AND HERZEGOVINA JAMAICA SIERRA LEONE BOTSWANA JAPAN BRAZIL JORDAN SINGAPORE BULGARIA KAZAKHSTAN SLOVAKIA BURKINA FASO KENYA SLOVENIA CAMEROON KOREA, REPUBLIC OF SOUTH AFRICA CANADA KUWAIT SPAIN CENTRAL AFRICAN KYRGYZSTAN SRI LANKA REPUBLIC LATVIA SUDAN CHAD LEBANON SWEDEN CHILE LIBERIA SWITZERLAND CHINA LIBYAN ARAB JAMAHIRIYA SYRIAN ARAB REPUBLIC COLOMBIA LIECHTENSTEIN TAJIKISTAN COSTA RICA LITHUANIA THAILAND CÔTE D’IVOIRE LUXEMBOURG THE FORMER YUGOSLAV CROATIA MADAGASCAR REPUBLIC OF MACEDONIA CUBA MALAYSIA TUNISIA CYPRUS MALI TURKEY CZECH REPUBLIC MALTA UGANDA DEMOCRATIC REPUBLIC MARSHALL ISLANDS UKRAINE OF THE CONGO MAURITANIA UNITED ARAB EMIRATES DENMARK MAURITIUS UNITED KINGDOM OF DOMINICAN REPUBLIC MEXICO GREAT BRITAIN AND ECUADOR MONACO NORTHERN IRELAND EGYPT MONGOLIA UNITED REPUBLIC EL SALVADOR MOROCCO ERITREA MYANMAR OF TANZANIA ESTONIA NAMIBIA UNITED STATES OF AMERICA ETHIOPIA NETHERLANDS URUGUAY FINLAND NEW ZEALAND UZBEKISTAN FRANCE NICARAGUA VENEZUELA GABON NIGER VIETNAM GEORGIA NIGERIA YEMEN GERMANY NORWAY ZAMBIA GHANA PAKISTAN ZIMBABWE The Agency’s Statute was approved on 23 October 1956 by the Conference on the Statute of the IAEA held at United Nations Headquarters, New York; it entered into force on 29 July 1957. The Headquarters of the Agency are situated in Vienna. -

A Study of Lithium Precursors on Nanoparticle Quality
Electronic Supplementary Material (ESI) for Nanoscale. This journal is © The Royal Society of Chemistry 2021 Electronic Supplementary Information Elucidating the role of precursors in synthesizing single crystalline lithium niobate nanomaterials: A study of lithium precursors on nanoparticle quality Rana Faryad Ali, Byron D. Gates* Department of Chemistry and 4D LABS, Simon Fraser University, 8888 University Drive Burnaby, BC, V5A 1S6, Canada * E-mail: [email protected] This work was supported in part by the Natural Sciences and Engineering Research Council of Canada (NSERC; Grant No. RGPIN-2020-06522), and through the Collaborative Health Research Projects (CHRP) Partnership Program supported in part by the Canadian Institutes of Health Research (Grant No. 134742) and the Natural Science Engineering Research Council of Canada (Grant No. CHRP 462260), the Canada Research Chairs Program (B.D. Gates, Grant No. 950-215846), CMC Microsystems (MNT Grant No. 6345), and a Graduate Fellowship (Rana Faryad Ali) from Simon Fraser University. This work made use of 4D LABS (www.4dlabs.com) and the Center for Soft Materials shared facilities supported by the Canada Foundation for Innovation (CFI), British Columbia Knowledge Development Fund (BCKDF), Western Economic Diversification Canada, and Simon Fraser University. S1 Experimental Materials and supplies All chemicals were of analytical grade and were used as received without further purification. Niobium ethoxide [Nb(OC2H5)5, >90%] was obtained from Gelest Inc., and benzyl alcohol (C7H7OH, 99%) and triethylamine [N(C2H5)3, 99.0%] were purchased from Acros Organics and Anachemia, respectively. Lithium chloride (LiCl, ~99.0%) was obtained from BDH Chemicals, and lithium bromide (LiBr, ≥99.0%), lithium fluoride (LiF, ~99.9%), and lithium iodide (LiI, 99.0%) were purchased from Sigma Aldrich. -

"Fluorine Compounds, Organic," In: Ullmann's Encyclopedia Of
Article No : a11_349 Fluorine Compounds, Organic GU¨ NTER SIEGEMUND, Hoechst Aktiengesellschaft, Frankfurt, Federal Republic of Germany WERNER SCHWERTFEGER, Hoechst Aktiengesellschaft, Frankfurt, Federal Republic of Germany ANDREW FEIRING, E. I. DuPont de Nemours & Co., Wilmington, Delaware, United States BRUCE SMART, E. I. DuPont de Nemours & Co., Wilmington, Delaware, United States FRED BEHR, Minnesota Mining and Manufacturing Company, St. Paul, Minnesota, United States HERWARD VOGEL, Minnesota Mining and Manufacturing Company, St. Paul, Minnesota, United States BLAINE MCKUSICK, E. I. DuPont de Nemours & Co., Wilmington, Delaware, United States 1. Introduction....................... 444 8. Fluorinated Carboxylic Acids and 2. Production Processes ................ 445 Fluorinated Alkanesulfonic Acids ...... 470 2.1. Substitution of Hydrogen............. 445 8.1. Fluorinated Carboxylic Acids ......... 470 2.2. Halogen – Fluorine Exchange ......... 446 8.1.1. Fluorinated Acetic Acids .............. 470 2.3. Synthesis from Fluorinated Synthons ... 447 8.1.2. Long-Chain Perfluorocarboxylic Acids .... 470 2.4. Addition of Hydrogen Fluoride to 8.1.3. Fluorinated Dicarboxylic Acids ......... 472 Unsaturated Bonds ................. 447 8.1.4. Tetrafluoroethylene – Perfluorovinyl Ether 2.5. Miscellaneous Methods .............. 447 Copolymers with Carboxylic Acid Groups . 472 2.6. Purification and Analysis ............. 447 8.2. Fluorinated Alkanesulfonic Acids ...... 472 3. Fluorinated Alkanes................. 448 8.2.1. Perfluoroalkanesulfonic Acids -

United States Patent Office Patented July 1, 1969
3,453,337 United States Patent Office Patented July 1, 1969 1. 2 3,453,337 FLUORINATION OF HALOGENATED The presence in the reaction mixture of the two fluo ORGANIC COMPOUNDS rides, or the complex fluoride enables better yields of Royston Henry Bennett and David Walter Cottrell, Ayon highly fluorinated products to be obtained under less mouth, England, assignors to Imperial Smelting Cor severe reaction condions, markedly increases the amount poration (N.S.C.) Limited, London, England, a British of fluorination reagent reacted under otherwise similar company conditions and enables the fluorination reaction to be No brawing. Filed Feb. 19, 1965, Ser. No. 434,128 carried out (for the same yield of product) at a lower Claims priority, application Great Britain, Feb. 26, 1964, temperature with the consequent use of less costly ma 7,932/64 terials and techniques of reactor construction. The pres Int, C. C07c 25/04 10 ence of the fluorides enables the vapor phase reaction U.S. C. 260-650 2 Claims to be carried out (for the same yields) at lower pressure This invention relates to the fluorination of organic than the pressure involved in the reactions using only halogen compounds and more especially to a process the alkali metal fluorides as proposed hitherto. The fur for the production of highly fluorinated aromatic com ther possibility of using a continuous flow apparatus such pounds by the replacement of higher halogen atoms in 15 as a fluidised reactor will be apparent to those familiar halogeno-aromatic compounds by fluorine atoms. with the art. The presence of the two fluorides or com Aromatic halogenocarbons containing carbon and halo plex fluoride enables a lower temperature to be em gen atoms only can be reacted with alkali fluorides ployed than was hitherto believed to be necessary, with under various conditions to give yields of halofluoro a consequent reduction in the extent of thermal degrada aromatic compounds. -

S41467-020-19206-W.Pdf
ARTICLE https://doi.org/10.1038/s41467-020-19206-w OPEN Unravelling the room-temperature atomic structure and growth kinetics of lithium metal Chao Liang 1, Xun Zhang1, Shuixin Xia1, Zeyu Wang1, Jiayi Wu1, Biao Yuan 1, Xin Luo1, Weiyan Liu1, ✉ Wei Liu 1 &YiYu 1 Alkali metals are widely studied in various fields such as medicine and battery. However, limited by the chemical reactivity and electron/ion beam sensitivity, the intrinsic atomic 1234567890():,; structure of alkali metals and its fundamental properties are difficult to be revealed. Here, a simple and versatile method is proposed to form the alkali metals in situ inside the transmission electron microscope. Taking alkali salts as the starting materials and electron beam as the trigger, alkali metals can be obtained directly. With this method, atomic resolution imaging of lithium and sodium metal is achieved at room temperature, and the growth of alkali metals is visualized at atomic-scale with millisecond temporal resolution. Furthermore, our observations unravel the ambiguities in lithium metal growth on garnet-type solid electrolytes for lithium-metal batteries. Finally, our method enables a direct study of physical contact property of lithium metal as well as its surface passivation oxide layer, which may contribute to better understanding of lithium dendrite and solid electrolyte interphase issues in lithium ion batteries. ✉ 1 School of Physical Science and Technology, ShanghaiTech University, Shanghai 201210, China. email: [email protected] NATURE COMMUNICATIONS | (2020) 11:5367 | https://doi.org/10.1038/s41467-020-19206-w | www.nature.com/naturecommunications 1 ARTICLE NATURE COMMUNICATIONS | https://doi.org/10.1038/s41467-020-19206-w t has been a long history for the research on alkali metals1, the to control the dose-rate of the electron beam. -

Pacs by Chemical Name (Mg/M3) (Pdf)
Table 4: Protective Action Criteria (PAC) Rev 25 based on applicable 60-minute AEGLs, ERPGs, or TEELs. Values are presented in mg/m3. August 2009 Table 4 is an alphabetical listing of the chemicals in the PAC data set. It provides Chemical Abstract Service Registry Numbers (CASRNs)1, PAC values, and technical information on the source of the PAC values. Table 4 presents all values for TEEL-0, PAC-1, PAC-2, and PAC-3 in mg/m3. The conversion of ppm to mg/m3 is calculated assuming 25 ºC and 760 mm Hg. The columns presented in Table 4 provide the following information: Heading Definition No. The ordered numbering of the chemicals as they appear in this alphabetical listing. Chemical Name The chemical name given to the PAC Development Team. CASRN The Chemical Abstract Service Registry Number for this chemical. TEEL-0 This is the threshold concentration below which most people will experience no adverse health effects. This PAC is always based on TEEL-0 because AEGL-0 or ERPG-0 values do not exist. PAC-1 Based on the applicable AEGL-1, ERPG-1, or TEEL-1 value. PAC-2 Based on the applicable AEGL-2, ERPG-2, or TEEL-2 value. PAC-3 Based on the applicable AEGL-3, ERPG-3, or TEEL-3 value. Source of PACs: Technical comments provided by the PAC development team that TEEL-0, PAC-1, indicate the source of the data used to derive PAC values. Future efforts PAC-2, PAC-3 are directed at reviewing, revising, and enhancing this information. -
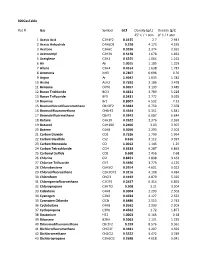
Gas Conversion Factor for 300 Series
300GasTable Rec # Gas Symbol GCF Density (g/L) Density (g/L) 25° C / 1 atm 0° C / 1 atm 1 Acetic Acid C2H4F2 0.4155 2.7 2.947 2 Acetic Anhydride C4H6O3 0.258 4.173 4.555 3 Acetone C3H6O 0.3556 2.374 2.591 4 Acetonitryl C2H3N 0.5178 1.678 1.832 5 Acetylene C2H2 0.6255 1.064 1.162 6 Air Air 1.0015 1.185 1.293 7 Allene C3H4 0.4514 1.638 1.787 8 Ammonia NH3 0.7807 0.696 0.76 9 Argon Ar 1.4047 1.633 1.782 10 Arsine AsH3 0.7592 3.186 3.478 11 Benzene C6H6 0.3057 3.193 3.485 12 Boron Trichloride BCl3 0.4421 4.789 5.228 13 Boron Triflouride BF3 0.5431 2.772 3.025 14 Bromine Br2 0.8007 6.532 7.13 15 Bromochlorodifluoromethane CBrClF2 0.3684 6.759 7.378 16 Bromodifluoromethane CHBrF2 0.4644 5.351 5.841 17 Bromotrifluormethane CBrF3 0.3943 6.087 6.644 18 Butane C4H10 0.2622 2.376 2.593 19 Butanol C4H10O 0.2406 3.03 3.307 20 Butene C4H8 0.3056 2.293 2.503 21 Carbon Dioxide CO2 0.7526 1.799 1.964 22 Carbon Disulfide CS2 0.616 3.112 3.397 23 Carbon Monoxide CO 1.0012 1.145 1.25 24 Carbon Tetrachloride CCl4 0.3333 6.287 6.863 25 Carbonyl Sulfide COS 0.668 2.456 2.68 26 Chlorine Cl2 0.8451 2.898 3.163 27 Chlorine Trifluoride ClF3 0.4496 3.779 4.125 28 Chlorobenzene C6H5Cl 0.2614 4.601 5.022 29 Chlorodifluoroethane C2H3ClF2 0.3216 4.108 4.484 30 Chloroform CHCl3 0.4192 4.879 5.326 31 Chloropentafluoroethane C2ClF5 0.2437 6.314 6.892 32 Chloropropane C3H7Cl 0.308 3.21 3.504 33 Cisbutene C4H8 0.3004 2.293 2.503 34 Cyanogen C2N2 0.4924 2.127 2.322 35 Cyanogen Chloride ClCN 0.6486 2.513 2.743 36 Cyclobutane C4H8 0.3562 2.293 2.503 37 Cyclopropane C3H6 0.4562 -

Ionic Conductivity of Solid Mixtures
NASA TECHNICAL NOTE NASA-- TN D-4606 d./ *o 0 *o nP LOAN COPY: RRURN TO AFWL (WLIL-2) KIRTLAND AFB, N MEX IONIC CONDUCTIVITY OF SOLID MIXTURES by William L. Fielder Lewis Research Celzter 3 ._i _... Cleveland, Ohio r I t , 1 NATIONAL AERONAUTICS AND SPACE ADMINISTRATION WASHINGTON, D. C. MAY 1968 ,/ I I TECH LIBRARY KAFB, NM ,lllllllllll__ .llllllllll I1111 lllllllllllllllll I 023L03b NASA TN D-4606 IONIC CONDUCTIVITY OF SOLID MIXTURES By William L. Fielder Lewis Research Center Cleveland, Ohio NATIONAL AERONAUTICS AND SPACE ADMINISTRATION For sale by the Clearinghouse for Federal Scientific and Technicol Information Springfield, Virginia 22151 - CFSTI price $3.00 IONIC CONDUCTIVITY OF SOLID MIXTURES by William L. Fielder Lewis Research Center SUMMARY The conductivities of four solid mixtures were determined as a function of tempera ture: (1) the lithium fluoride - lithium chloride eutectic, (2) the lithium chloride - potassium chloride eutectic, (3) the lithium fluoride - sodium chloride eutectic, and (4) a 50-mole-percent mixture of sodium chloride and potassium chloride. Two conductivity regions were obtained for each of the four mixtures. The activation energies for the conductivity for the lower-temperature regions ranged from 14 to 27 kilocalories per mole (59 to 114 kJ/mole). These energies were similar to the cation migration energies for the single crystals of the alkali halides. The conductivity of the mixtures in the lower-temperature regions is best explained by the following mechanism: (1) formation of cation vacancies primarily by multivalent impurities, and (2) migration of the cations through these vacancies. The activation energies for the conductivity of the solid mixtures in the upper- temperature regions ranged from 5 to 9 kilocalories per mole (20 to 39 kJ/mole). -

Synthesis of Hexafluorobenzene Through Batch Reactive Distillation
SYNTHESIS OF HEXAFLUOROBENZENE THROUGH BATCH REACTIVE DISTILLATION Jesita Reddy [Bsc. Eng.] In fulfilment of the requirements for the degree Master of Science in Engineering, College of Agriculture, Engineering and Science, University of KwaZulu-Natal December 2018 Supervisors: Doctor David Lokhat and Professor Deresh Ramjugernath As the candidate’s supervisor I agree to the submission of this thesis: Dr. D. Lokhat Prof. D. Ramjugernath DECLARATION I, Jesita Reddy declare that: The research reported in this dissertation/thesis, except where otherwise indicated, is my original work. 1. The research reported in this thesis, except where otherwise indicated, is my original research. 2. This thesis has not been submitted for any degree or examination at any other university. 3. This thesis does not contain other persons’ data, pictures, graphs or other information, unless specifically acknowledged as being sourced from other persons. 4. This thesis does not contain other persons' writing, unless specifically acknowledged as being sourced from other researchers. Where other written sources have been quoted, then: i. Their words have been re-written but the general information attributed to them has been referenced ii. Where their exact words have been used, then their writing has been placed in italics and inside quotation marks, and referenced. 5. This thesis does not contain text, graphics or tables copied and pasted from the Internet, unless specifically acknowledged, and the source being detailed in the thesis and in the References sections. Signed: ________________ Date: ____13/03/19____________ ii ACKNOWLEDGEMENTS This research was affiliated with the Fluorochemical Expansion Initiative recognized by the Department of Science and Technology. -

Synthesis and Structure-Property Studies of Organic Materials Containing Fluorinated and Non-Fluorinated # Systems (Small Molecules and Polymers)" (2008)
University of Kentucky UKnowledge University of Kentucky Doctoral Dissertations Graduate School 2008 SYNTHESIS AND STRUCTURE-PROPERTY STUDIES OF ORGANIC MATERIALS CONTAINING FLUORINATED AND NON- FLUORINATED # SYSTEMS (SMALL MOLECULES AND POLYMERS) Yongfeng Wang University of Kentucky, [email protected] Right click to open a feedback form in a new tab to let us know how this document benefits ou.y Recommended Citation Wang, Yongfeng, "SYNTHESIS AND STRUCTURE-PROPERTY STUDIES OF ORGANIC MATERIALS CONTAINING FLUORINATED AND NON-FLUORINATED # SYSTEMS (SMALL MOLECULES AND POLYMERS)" (2008). University of Kentucky Doctoral Dissertations. 593. https://uknowledge.uky.edu/gradschool_diss/593 This Dissertation is brought to you for free and open access by the Graduate School at UKnowledge. It has been accepted for inclusion in University of Kentucky Doctoral Dissertations by an authorized administrator of UKnowledge. For more information, please contact [email protected]. ABSTRACT OF DISSERTATION Yongfeng Wang The Graduate School University of Kentucky 2008 SYNTHESIS AND STRUCTURE-PROPERTY STUDIES OF ORGANIC MATERIALS CONTAINING FLUORINATED AND NON-FLUORINATED π SYSTEMS (SMALL MOLECULES AND POLYMERS) ABSTRACT OF DISSERTATION A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the College of Arts and Science at the University of Kentucky By Yongfeng Wang Lexington, Kentucky Director: Dr. Mark D. Watson, Professor of Chemistry Lexington, Kentucky 2008 Copyright © Yongfeng Wang 2008 ABSTRACT OF DISSERTATION SYNTHESIS AND STRUCTURE-PROPERTY STUDIES OF ORGANIC MATERIALS CONTAINING FLUORINATED AND NON-FLUORINATED π SYSTEMS (SMALL MOLECULES AND POLYMERS) Organic electronic materials have high potential in consumer electronics. Challenges facing materials chemists include development of efficient synthetic methodologies and control over interrelated molecular self-assembly and (opto) electronic properties. -

Tetramethylammonium Fluoride Tetrahydrate for Snar Fluorination of 4‑Chlorothiazoles at a Production Scale Mai Khanh Hawk,* Sarah J
pubs.acs.org/OPRD Article Tetramethylammonium Fluoride Tetrahydrate for SNAr Fluorination of 4‑Chlorothiazoles at a Production Scale Mai Khanh Hawk,* Sarah J. Ryan,* Xin Zhang, Ping Huang, Jing Chen, Chuanren Liu, Jianping Chen, Peter J. Lindsay-Scott, John Burnett, Craig White, Yu Lu, and John R. Rizzo Cite This: https://doi.org/10.1021/acs.oprd.1c00042 Read Online ACCESS Metrics & More Article Recommendations *sı Supporting Information fl · ABSTRACT: This article describes the use of tetramethylammonium uoride tetrahydrate (TMAF 4H2O) for the large-scale fl · preparation of a challenging 4- uorothiazole. Commercially available TMAF 4H2O was procured on a large scale and rigorously dried by distillation with isopropyl alcohol and then dimethylformamide at elevated temperature. This method of drying provided anhydrous TMAF [TMAF (anh)] containing <0.2 wt % water and <60 ppm isopropanol. The use of TMAF (anh) was essential for production of the 4-fluorothiazole. When the chlorothiazole starting material was treated with other anhydrous fluoride sources, fl · poor conversion of the starting material or potential safety issues were observed. SNAr uorination using dried TMAF 4H2O was carried out at a 45.1 kg scale at 95−100 °C to produce 36.8 kg of 4-fluorothiazole 1b. fl fl fl fl KEYWORDS: uorination, SNAr, tetramethylammonium uoride, uorothiazole, heteroarene, anhydrous uoride ■ INTRODUCTION demonstrated that anhydrous tetrabutylammonium fluoride fl [TBAF (anh)] could be prepared in situ from tetrabutylam- The substitution of hydrogen for uorine can result in fl monium cyanide (TBACN) and hexa uorobenzene (C6F6) improved bioavailability or metabolic stability of bioactive 11 1 (Scheme 2A). This reagent was used for the room- molecules.