Synthesis of Hexafluorobenzene Through Batch Reactive Distillation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Durham E-Theses
Durham E-Theses Halogenated diazines and triazines Wood, D. E. How to cite: Wood, D. E. (1978) Halogenated diazines and triazines, Durham theses, Durham University. Available at Durham E-Theses Online: http://etheses.dur.ac.uk/8324/ Use policy The full-text may be used and/or reproduced, and given to third parties in any format or medium, without prior permission or charge, for personal research or study, educational, or not-for-prot purposes provided that: • a full bibliographic reference is made to the original source • a link is made to the metadata record in Durham E-Theses • the full-text is not changed in any way The full-text must not be sold in any format or medium without the formal permission of the copyright holders. Please consult the full Durham E-Theses policy for further details. Academic Support Oce, Durham University, University Oce, Old Elvet, Durham DH1 3HP e-mail: [email protected] Tel: +44 0191 334 6107 http://etheses.dur.ac.uk UNIVERSITY OF Du'RKAM A THESIS entitled HALOGENATED DIAZINES AND TRIAZINES Submitted by D E. WOOD (Grey), B Sc (London) The copyright of this thesis rests with the author No quotation from it should be published without his prior written consent and information derived from it should be acknowledged A candidate for the degree of Doctor of Philosophy 19 78 sr i i > j J To my MoLhcr and FaLhcr WLLII Lh.inks for .ill LhaL Lhey have done ACKNOWLEDGEMLNTS I would like LO express my thanks Lo Professor R D Chambers i under whose guidance this research was undertaken, for considerable encouragement, advice and discussion Thanks are due to Dr R S Matthews for his expert advice with n in r. -

Use of Chlorofluorocarbons in Hydrology : a Guidebook
USE OF CHLOROFLUOROCARBONS IN HYDROLOGY A Guidebook USE OF CHLOROFLUOROCARBONS IN HYDROLOGY A GUIDEBOOK 2005 Edition The following States are Members of the International Atomic Energy Agency: AFGHANISTAN GREECE PANAMA ALBANIA GUATEMALA PARAGUAY ALGERIA HAITI PERU ANGOLA HOLY SEE PHILIPPINES ARGENTINA HONDURAS POLAND ARMENIA HUNGARY PORTUGAL AUSTRALIA ICELAND QATAR AUSTRIA INDIA REPUBLIC OF MOLDOVA AZERBAIJAN INDONESIA ROMANIA BANGLADESH IRAN, ISLAMIC REPUBLIC OF RUSSIAN FEDERATION BELARUS IRAQ SAUDI ARABIA BELGIUM IRELAND SENEGAL BENIN ISRAEL SERBIA AND MONTENEGRO BOLIVIA ITALY SEYCHELLES BOSNIA AND HERZEGOVINA JAMAICA SIERRA LEONE BOTSWANA JAPAN BRAZIL JORDAN SINGAPORE BULGARIA KAZAKHSTAN SLOVAKIA BURKINA FASO KENYA SLOVENIA CAMEROON KOREA, REPUBLIC OF SOUTH AFRICA CANADA KUWAIT SPAIN CENTRAL AFRICAN KYRGYZSTAN SRI LANKA REPUBLIC LATVIA SUDAN CHAD LEBANON SWEDEN CHILE LIBERIA SWITZERLAND CHINA LIBYAN ARAB JAMAHIRIYA SYRIAN ARAB REPUBLIC COLOMBIA LIECHTENSTEIN TAJIKISTAN COSTA RICA LITHUANIA THAILAND CÔTE D’IVOIRE LUXEMBOURG THE FORMER YUGOSLAV CROATIA MADAGASCAR REPUBLIC OF MACEDONIA CUBA MALAYSIA TUNISIA CYPRUS MALI TURKEY CZECH REPUBLIC MALTA UGANDA DEMOCRATIC REPUBLIC MARSHALL ISLANDS UKRAINE OF THE CONGO MAURITANIA UNITED ARAB EMIRATES DENMARK MAURITIUS UNITED KINGDOM OF DOMINICAN REPUBLIC MEXICO GREAT BRITAIN AND ECUADOR MONACO NORTHERN IRELAND EGYPT MONGOLIA UNITED REPUBLIC EL SALVADOR MOROCCO ERITREA MYANMAR OF TANZANIA ESTONIA NAMIBIA UNITED STATES OF AMERICA ETHIOPIA NETHERLANDS URUGUAY FINLAND NEW ZEALAND UZBEKISTAN FRANCE NICARAGUA VENEZUELA GABON NIGER VIETNAM GEORGIA NIGERIA YEMEN GERMANY NORWAY ZAMBIA GHANA PAKISTAN ZIMBABWE The Agency’s Statute was approved on 23 October 1956 by the Conference on the Statute of the IAEA held at United Nations Headquarters, New York; it entered into force on 29 July 1957. The Headquarters of the Agency are situated in Vienna. -

1 Abietic Acid R Abrasive Silica for Polishing DR Acenaphthene M (LC
1 abietic acid R abrasive silica for polishing DR acenaphthene M (LC) acenaphthene quinone R acenaphthylene R acetal (see 1,1-diethoxyethane) acetaldehyde M (FC) acetaldehyde-d (CH3CDO) R acetaldehyde dimethyl acetal CH acetaldoxime R acetamide M (LC) acetamidinium chloride R acetamidoacrylic acid 2- NB acetamidobenzaldehyde p- R acetamidobenzenesulfonyl chloride 4- R acetamidodeoxythioglucopyranose triacetate 2- -2- -1- -β-D- 3,4,6- AB acetamidomethylthiazole 2- -4- PB acetanilide M (LC) acetazolamide R acetdimethylamide see dimethylacetamide, N,N- acethydrazide R acetic acid M (solv) acetic anhydride M (FC) acetmethylamide see methylacetamide, N- acetoacetamide R acetoacetanilide R acetoacetic acid, lithium salt R acetobromoglucose -α-D- NB acetohydroxamic acid R acetoin R acetol (hydroxyacetone) R acetonaphthalide (α)R acetone M (solv) acetone ,A.R. M (solv) acetone-d6 RM acetone cyanohydrin R acetonedicarboxylic acid ,dimethyl ester R acetonedicarboxylic acid -1,3- R acetone dimethyl acetal see dimethoxypropane 2,2- acetonitrile M (solv) acetonitrile-d3 RM acetonylacetone see hexanedione 2,5- acetonylbenzylhydroxycoumarin (3-(α- -4- R acetophenone M (LC) acetophenone oxime R acetophenone trimethylsilyl enol ether see phenyltrimethylsilyl... acetoxyacetone (oxopropyl acetate 2-) R acetoxybenzoic acid 4- DS acetoxynaphthoic acid 6- -2- R 2 acetylacetaldehyde dimethylacetal R acetylacetone (pentanedione -2,4-) M (C) acetylbenzonitrile p- R acetylbiphenyl 4- see phenylacetophenone, p- acetyl bromide M (FC) acetylbromothiophene 2- -5- -

Novel Approaches Towards the Discovery of Tumor-Selective Histone Deacetylase Inhibitors
Novel Approaches Towards The Discovery of Tumor-Selective Histone Deacetylase Inhibitors A Dissertation Presented to The Academic Faculty By Idris Raji In Partial Fulfilment of the Requirements for the Degree Doctor of Philosophy in Chemistry School of Chemistry and Biochemistry Georgia Institute of Technology, Atlanta GA 30332 December 2016 COPYRIGHT 2016 © IDRIS RAJI Novel Approaches Towards The Discovery of Tumor-Selective Histone Deacetylase Inhibitors Committee members: Dr. Adegboyega K. Oyelere, Advisor Dr. Andreas Bommarius School of Chemistry and Biochemistry School of Chemical and Biomolecular Georgia Institute of Technology Engineering Georgia Institute of Technology Dr. M. G. Finn School of Chemistry and Biochemistry Dr. Ravi Bellamkonda Georgia Institute of Technology Pratt’s school of Engineering Duke University Dr. Stefan France School of Chemistry and Biochemistry Georgia Institute of Technology Date Approved: November 4th, 2016 To my mother, Mrs. Jolade Raji AKNOWLEDGEMENTS I have been very fortunate to have met and interact with people who have positively influenced my life since arriving at Georgia Tech for graduate studies. I will be forever grateful to my advisor, Dr. Adegboyega K. Oyelere, for giving me the opportunity to learn in his lab. I joined his lab with minimal research experience, but he has helped to tremendously enrich my knowledge base and instill a level of confidence in me that was previously nonexistent. I am very grateful for his patience, guidance, availability to discuss research progress and the countless recommendation letters he wrote on my behalf. I am greatly indebted to my thesis committee members Dr. M G Finn, Dr. Stefan France, Dr. Ravi Bellamkonda, and Dr. -

"Fluorine Compounds, Organic," In: Ullmann's Encyclopedia Of
Article No : a11_349 Fluorine Compounds, Organic GU¨ NTER SIEGEMUND, Hoechst Aktiengesellschaft, Frankfurt, Federal Republic of Germany WERNER SCHWERTFEGER, Hoechst Aktiengesellschaft, Frankfurt, Federal Republic of Germany ANDREW FEIRING, E. I. DuPont de Nemours & Co., Wilmington, Delaware, United States BRUCE SMART, E. I. DuPont de Nemours & Co., Wilmington, Delaware, United States FRED BEHR, Minnesota Mining and Manufacturing Company, St. Paul, Minnesota, United States HERWARD VOGEL, Minnesota Mining and Manufacturing Company, St. Paul, Minnesota, United States BLAINE MCKUSICK, E. I. DuPont de Nemours & Co., Wilmington, Delaware, United States 1. Introduction....................... 444 8. Fluorinated Carboxylic Acids and 2. Production Processes ................ 445 Fluorinated Alkanesulfonic Acids ...... 470 2.1. Substitution of Hydrogen............. 445 8.1. Fluorinated Carboxylic Acids ......... 470 2.2. Halogen – Fluorine Exchange ......... 446 8.1.1. Fluorinated Acetic Acids .............. 470 2.3. Synthesis from Fluorinated Synthons ... 447 8.1.2. Long-Chain Perfluorocarboxylic Acids .... 470 2.4. Addition of Hydrogen Fluoride to 8.1.3. Fluorinated Dicarboxylic Acids ......... 472 Unsaturated Bonds ................. 447 8.1.4. Tetrafluoroethylene – Perfluorovinyl Ether 2.5. Miscellaneous Methods .............. 447 Copolymers with Carboxylic Acid Groups . 472 2.6. Purification and Analysis ............. 447 8.2. Fluorinated Alkanesulfonic Acids ...... 472 3. Fluorinated Alkanes................. 448 8.2.1. Perfluoroalkanesulfonic Acids -

United States Patent Office Patented July 1, 1969
3,453,337 United States Patent Office Patented July 1, 1969 1. 2 3,453,337 FLUORINATION OF HALOGENATED The presence in the reaction mixture of the two fluo ORGANIC COMPOUNDS rides, or the complex fluoride enables better yields of Royston Henry Bennett and David Walter Cottrell, Ayon highly fluorinated products to be obtained under less mouth, England, assignors to Imperial Smelting Cor severe reaction condions, markedly increases the amount poration (N.S.C.) Limited, London, England, a British of fluorination reagent reacted under otherwise similar company conditions and enables the fluorination reaction to be No brawing. Filed Feb. 19, 1965, Ser. No. 434,128 carried out (for the same yield of product) at a lower Claims priority, application Great Britain, Feb. 26, 1964, temperature with the consequent use of less costly ma 7,932/64 terials and techniques of reactor construction. The pres Int, C. C07c 25/04 10 ence of the fluorides enables the vapor phase reaction U.S. C. 260-650 2 Claims to be carried out (for the same yields) at lower pressure This invention relates to the fluorination of organic than the pressure involved in the reactions using only halogen compounds and more especially to a process the alkali metal fluorides as proposed hitherto. The fur for the production of highly fluorinated aromatic com ther possibility of using a continuous flow apparatus such pounds by the replacement of higher halogen atoms in 15 as a fluidised reactor will be apparent to those familiar halogeno-aromatic compounds by fluorine atoms. with the art. The presence of the two fluorides or com Aromatic halogenocarbons containing carbon and halo plex fluoride enables a lower temperature to be em gen atoms only can be reacted with alkali fluorides ployed than was hitherto believed to be necessary, with under various conditions to give yields of halofluoro a consequent reduction in the extent of thermal degrada aromatic compounds. -

Pacs by Chemical Name (Mg/M3) (Pdf)
Table 4: Protective Action Criteria (PAC) Rev 25 based on applicable 60-minute AEGLs, ERPGs, or TEELs. Values are presented in mg/m3. August 2009 Table 4 is an alphabetical listing of the chemicals in the PAC data set. It provides Chemical Abstract Service Registry Numbers (CASRNs)1, PAC values, and technical information on the source of the PAC values. Table 4 presents all values for TEEL-0, PAC-1, PAC-2, and PAC-3 in mg/m3. The conversion of ppm to mg/m3 is calculated assuming 25 ºC and 760 mm Hg. The columns presented in Table 4 provide the following information: Heading Definition No. The ordered numbering of the chemicals as they appear in this alphabetical listing. Chemical Name The chemical name given to the PAC Development Team. CASRN The Chemical Abstract Service Registry Number for this chemical. TEEL-0 This is the threshold concentration below which most people will experience no adverse health effects. This PAC is always based on TEEL-0 because AEGL-0 or ERPG-0 values do not exist. PAC-1 Based on the applicable AEGL-1, ERPG-1, or TEEL-1 value. PAC-2 Based on the applicable AEGL-2, ERPG-2, or TEEL-2 value. PAC-3 Based on the applicable AEGL-3, ERPG-3, or TEEL-3 value. Source of PACs: Technical comments provided by the PAC development team that TEEL-0, PAC-1, indicate the source of the data used to derive PAC values. Future efforts PAC-2, PAC-3 are directed at reviewing, revising, and enhancing this information. -

WO 2016/074683 Al 19 May 2016 (19.05.2016) W P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/074683 Al 19 May 2016 (19.05.2016) W P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every C12N 15/10 (2006.01) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (21) International Application Number: BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, PCT/DK20 15/050343 DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (22) International Filing Date: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, 11 November 2015 ( 11. 1 1.2015) KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (25) Filing Language: English PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, (26) Publication Language: English SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: PA 2014 00655 11 November 2014 ( 11. 1 1.2014) DK (84) Designated States (unless otherwise indicated, for every 62/077,933 11 November 2014 ( 11. 11.2014) US kind of regional protection available): ARIPO (BW, GH, 62/202,3 18 7 August 2015 (07.08.2015) US GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, (71) Applicant: LUNDORF PEDERSEN MATERIALS APS TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, [DK/DK]; Nordvej 16 B, Himmelev, DK-4000 Roskilde DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, (DK). -
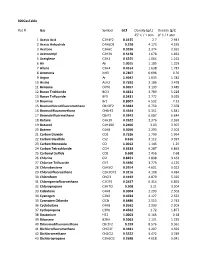
Gas Conversion Factor for 300 Series
300GasTable Rec # Gas Symbol GCF Density (g/L) Density (g/L) 25° C / 1 atm 0° C / 1 atm 1 Acetic Acid C2H4F2 0.4155 2.7 2.947 2 Acetic Anhydride C4H6O3 0.258 4.173 4.555 3 Acetone C3H6O 0.3556 2.374 2.591 4 Acetonitryl C2H3N 0.5178 1.678 1.832 5 Acetylene C2H2 0.6255 1.064 1.162 6 Air Air 1.0015 1.185 1.293 7 Allene C3H4 0.4514 1.638 1.787 8 Ammonia NH3 0.7807 0.696 0.76 9 Argon Ar 1.4047 1.633 1.782 10 Arsine AsH3 0.7592 3.186 3.478 11 Benzene C6H6 0.3057 3.193 3.485 12 Boron Trichloride BCl3 0.4421 4.789 5.228 13 Boron Triflouride BF3 0.5431 2.772 3.025 14 Bromine Br2 0.8007 6.532 7.13 15 Bromochlorodifluoromethane CBrClF2 0.3684 6.759 7.378 16 Bromodifluoromethane CHBrF2 0.4644 5.351 5.841 17 Bromotrifluormethane CBrF3 0.3943 6.087 6.644 18 Butane C4H10 0.2622 2.376 2.593 19 Butanol C4H10O 0.2406 3.03 3.307 20 Butene C4H8 0.3056 2.293 2.503 21 Carbon Dioxide CO2 0.7526 1.799 1.964 22 Carbon Disulfide CS2 0.616 3.112 3.397 23 Carbon Monoxide CO 1.0012 1.145 1.25 24 Carbon Tetrachloride CCl4 0.3333 6.287 6.863 25 Carbonyl Sulfide COS 0.668 2.456 2.68 26 Chlorine Cl2 0.8451 2.898 3.163 27 Chlorine Trifluoride ClF3 0.4496 3.779 4.125 28 Chlorobenzene C6H5Cl 0.2614 4.601 5.022 29 Chlorodifluoroethane C2H3ClF2 0.3216 4.108 4.484 30 Chloroform CHCl3 0.4192 4.879 5.326 31 Chloropentafluoroethane C2ClF5 0.2437 6.314 6.892 32 Chloropropane C3H7Cl 0.308 3.21 3.504 33 Cisbutene C4H8 0.3004 2.293 2.503 34 Cyanogen C2N2 0.4924 2.127 2.322 35 Cyanogen Chloride ClCN 0.6486 2.513 2.743 36 Cyclobutane C4H8 0.3562 2.293 2.503 37 Cyclopropane C3H6 0.4562 -

Downloaded for Personal Non-Commercial Research Or Study, Without Prior Permission Or Charge
https://theses.gla.ac.uk/ Theses Digitisation: https://www.gla.ac.uk/myglasgow/research/enlighten/theses/digitisation/ This is a digitised version of the original print thesis. Copyright and moral rights for this work are retained by the author A copy can be downloaded for personal non-commercial research or study, without prior permission or charge This work cannot be reproduced or quoted extensively from without first obtaining permission in writing from the author The content must not be changed in any way or sold commercially in any format or medium without the formal permission of the author When referring to this work, full bibliographic details including the author, title, awarding institution and date of the thesis must be given Enlighten: Theses https://theses.gla.ac.uk/ [email protected] RADIOTRACER STUDIES OF THE CHLOROFLUORINATION OF SULPHUR TETRAFLUORIDE AT MERCURY(II) FLUORIDE Thesis submitted to the University of Glasgow for the degree of M.Sc BY MOHAMED SELOUGHA FACULTY OF SCIENCE DEPARTMENT OF CHEMISTRY SEPTEMBER 1990 ProQuest Number: 11007548 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a com plete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest ProQuest 11007548 Published by ProQuest LLC(2018). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States C ode Microform Edition © ProQuest LLC. -

Synthesis and Structure-Property Studies of Organic Materials Containing Fluorinated and Non-Fluorinated # Systems (Small Molecules and Polymers)" (2008)
University of Kentucky UKnowledge University of Kentucky Doctoral Dissertations Graduate School 2008 SYNTHESIS AND STRUCTURE-PROPERTY STUDIES OF ORGANIC MATERIALS CONTAINING FLUORINATED AND NON- FLUORINATED # SYSTEMS (SMALL MOLECULES AND POLYMERS) Yongfeng Wang University of Kentucky, [email protected] Right click to open a feedback form in a new tab to let us know how this document benefits ou.y Recommended Citation Wang, Yongfeng, "SYNTHESIS AND STRUCTURE-PROPERTY STUDIES OF ORGANIC MATERIALS CONTAINING FLUORINATED AND NON-FLUORINATED # SYSTEMS (SMALL MOLECULES AND POLYMERS)" (2008). University of Kentucky Doctoral Dissertations. 593. https://uknowledge.uky.edu/gradschool_diss/593 This Dissertation is brought to you for free and open access by the Graduate School at UKnowledge. It has been accepted for inclusion in University of Kentucky Doctoral Dissertations by an authorized administrator of UKnowledge. For more information, please contact [email protected]. ABSTRACT OF DISSERTATION Yongfeng Wang The Graduate School University of Kentucky 2008 SYNTHESIS AND STRUCTURE-PROPERTY STUDIES OF ORGANIC MATERIALS CONTAINING FLUORINATED AND NON-FLUORINATED π SYSTEMS (SMALL MOLECULES AND POLYMERS) ABSTRACT OF DISSERTATION A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the College of Arts and Science at the University of Kentucky By Yongfeng Wang Lexington, Kentucky Director: Dr. Mark D. Watson, Professor of Chemistry Lexington, Kentucky 2008 Copyright © Yongfeng Wang 2008 ABSTRACT OF DISSERTATION SYNTHESIS AND STRUCTURE-PROPERTY STUDIES OF ORGANIC MATERIALS CONTAINING FLUORINATED AND NON-FLUORINATED π SYSTEMS (SMALL MOLECULES AND POLYMERS) Organic electronic materials have high potential in consumer electronics. Challenges facing materials chemists include development of efficient synthetic methodologies and control over interrelated molecular self-assembly and (opto) electronic properties. -

Tetramethylammonium Fluoride Tetrahydrate for Snar Fluorination of 4‑Chlorothiazoles at a Production Scale Mai Khanh Hawk,* Sarah J
pubs.acs.org/OPRD Article Tetramethylammonium Fluoride Tetrahydrate for SNAr Fluorination of 4‑Chlorothiazoles at a Production Scale Mai Khanh Hawk,* Sarah J. Ryan,* Xin Zhang, Ping Huang, Jing Chen, Chuanren Liu, Jianping Chen, Peter J. Lindsay-Scott, John Burnett, Craig White, Yu Lu, and John R. Rizzo Cite This: https://doi.org/10.1021/acs.oprd.1c00042 Read Online ACCESS Metrics & More Article Recommendations *sı Supporting Information fl · ABSTRACT: This article describes the use of tetramethylammonium uoride tetrahydrate (TMAF 4H2O) for the large-scale fl · preparation of a challenging 4- uorothiazole. Commercially available TMAF 4H2O was procured on a large scale and rigorously dried by distillation with isopropyl alcohol and then dimethylformamide at elevated temperature. This method of drying provided anhydrous TMAF [TMAF (anh)] containing <0.2 wt % water and <60 ppm isopropanol. The use of TMAF (anh) was essential for production of the 4-fluorothiazole. When the chlorothiazole starting material was treated with other anhydrous fluoride sources, fl · poor conversion of the starting material or potential safety issues were observed. SNAr uorination using dried TMAF 4H2O was carried out at a 45.1 kg scale at 95−100 °C to produce 36.8 kg of 4-fluorothiazole 1b. fl fl fl fl KEYWORDS: uorination, SNAr, tetramethylammonium uoride, uorothiazole, heteroarene, anhydrous uoride ■ INTRODUCTION demonstrated that anhydrous tetrabutylammonium fluoride fl [TBAF (anh)] could be prepared in situ from tetrabutylam- The substitution of hydrogen for uorine can result in fl monium cyanide (TBACN) and hexa uorobenzene (C6F6) improved bioavailability or metabolic stability of bioactive 11 1 (Scheme 2A). This reagent was used for the room- molecules.