Synthesis and Structure-Property Studies of Organic Materials Containing Fluorinated and Non-Fluorinated # Systems (Small Molecules and Polymers)" (2008)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Use of Chlorofluorocarbons in Hydrology : a Guidebook
USE OF CHLOROFLUOROCARBONS IN HYDROLOGY A Guidebook USE OF CHLOROFLUOROCARBONS IN HYDROLOGY A GUIDEBOOK 2005 Edition The following States are Members of the International Atomic Energy Agency: AFGHANISTAN GREECE PANAMA ALBANIA GUATEMALA PARAGUAY ALGERIA HAITI PERU ANGOLA HOLY SEE PHILIPPINES ARGENTINA HONDURAS POLAND ARMENIA HUNGARY PORTUGAL AUSTRALIA ICELAND QATAR AUSTRIA INDIA REPUBLIC OF MOLDOVA AZERBAIJAN INDONESIA ROMANIA BANGLADESH IRAN, ISLAMIC REPUBLIC OF RUSSIAN FEDERATION BELARUS IRAQ SAUDI ARABIA BELGIUM IRELAND SENEGAL BENIN ISRAEL SERBIA AND MONTENEGRO BOLIVIA ITALY SEYCHELLES BOSNIA AND HERZEGOVINA JAMAICA SIERRA LEONE BOTSWANA JAPAN BRAZIL JORDAN SINGAPORE BULGARIA KAZAKHSTAN SLOVAKIA BURKINA FASO KENYA SLOVENIA CAMEROON KOREA, REPUBLIC OF SOUTH AFRICA CANADA KUWAIT SPAIN CENTRAL AFRICAN KYRGYZSTAN SRI LANKA REPUBLIC LATVIA SUDAN CHAD LEBANON SWEDEN CHILE LIBERIA SWITZERLAND CHINA LIBYAN ARAB JAMAHIRIYA SYRIAN ARAB REPUBLIC COLOMBIA LIECHTENSTEIN TAJIKISTAN COSTA RICA LITHUANIA THAILAND CÔTE D’IVOIRE LUXEMBOURG THE FORMER YUGOSLAV CROATIA MADAGASCAR REPUBLIC OF MACEDONIA CUBA MALAYSIA TUNISIA CYPRUS MALI TURKEY CZECH REPUBLIC MALTA UGANDA DEMOCRATIC REPUBLIC MARSHALL ISLANDS UKRAINE OF THE CONGO MAURITANIA UNITED ARAB EMIRATES DENMARK MAURITIUS UNITED KINGDOM OF DOMINICAN REPUBLIC MEXICO GREAT BRITAIN AND ECUADOR MONACO NORTHERN IRELAND EGYPT MONGOLIA UNITED REPUBLIC EL SALVADOR MOROCCO ERITREA MYANMAR OF TANZANIA ESTONIA NAMIBIA UNITED STATES OF AMERICA ETHIOPIA NETHERLANDS URUGUAY FINLAND NEW ZEALAND UZBEKISTAN FRANCE NICARAGUA VENEZUELA GABON NIGER VIETNAM GEORGIA NIGERIA YEMEN GERMANY NORWAY ZAMBIA GHANA PAKISTAN ZIMBABWE The Agency’s Statute was approved on 23 October 1956 by the Conference on the Statute of the IAEA held at United Nations Headquarters, New York; it entered into force on 29 July 1957. The Headquarters of the Agency are situated in Vienna. -

"Fluorine Compounds, Organic," In: Ullmann's Encyclopedia Of
Article No : a11_349 Fluorine Compounds, Organic GU¨ NTER SIEGEMUND, Hoechst Aktiengesellschaft, Frankfurt, Federal Republic of Germany WERNER SCHWERTFEGER, Hoechst Aktiengesellschaft, Frankfurt, Federal Republic of Germany ANDREW FEIRING, E. I. DuPont de Nemours & Co., Wilmington, Delaware, United States BRUCE SMART, E. I. DuPont de Nemours & Co., Wilmington, Delaware, United States FRED BEHR, Minnesota Mining and Manufacturing Company, St. Paul, Minnesota, United States HERWARD VOGEL, Minnesota Mining and Manufacturing Company, St. Paul, Minnesota, United States BLAINE MCKUSICK, E. I. DuPont de Nemours & Co., Wilmington, Delaware, United States 1. Introduction....................... 444 8. Fluorinated Carboxylic Acids and 2. Production Processes ................ 445 Fluorinated Alkanesulfonic Acids ...... 470 2.1. Substitution of Hydrogen............. 445 8.1. Fluorinated Carboxylic Acids ......... 470 2.2. Halogen – Fluorine Exchange ......... 446 8.1.1. Fluorinated Acetic Acids .............. 470 2.3. Synthesis from Fluorinated Synthons ... 447 8.1.2. Long-Chain Perfluorocarboxylic Acids .... 470 2.4. Addition of Hydrogen Fluoride to 8.1.3. Fluorinated Dicarboxylic Acids ......... 472 Unsaturated Bonds ................. 447 8.1.4. Tetrafluoroethylene – Perfluorovinyl Ether 2.5. Miscellaneous Methods .............. 447 Copolymers with Carboxylic Acid Groups . 472 2.6. Purification and Analysis ............. 447 8.2. Fluorinated Alkanesulfonic Acids ...... 472 3. Fluorinated Alkanes................. 448 8.2.1. Perfluoroalkanesulfonic Acids -

United States Patent Office Patented July 1, 1969
3,453,337 United States Patent Office Patented July 1, 1969 1. 2 3,453,337 FLUORINATION OF HALOGENATED The presence in the reaction mixture of the two fluo ORGANIC COMPOUNDS rides, or the complex fluoride enables better yields of Royston Henry Bennett and David Walter Cottrell, Ayon highly fluorinated products to be obtained under less mouth, England, assignors to Imperial Smelting Cor severe reaction condions, markedly increases the amount poration (N.S.C.) Limited, London, England, a British of fluorination reagent reacted under otherwise similar company conditions and enables the fluorination reaction to be No brawing. Filed Feb. 19, 1965, Ser. No. 434,128 carried out (for the same yield of product) at a lower Claims priority, application Great Britain, Feb. 26, 1964, temperature with the consequent use of less costly ma 7,932/64 terials and techniques of reactor construction. The pres Int, C. C07c 25/04 10 ence of the fluorides enables the vapor phase reaction U.S. C. 260-650 2 Claims to be carried out (for the same yields) at lower pressure This invention relates to the fluorination of organic than the pressure involved in the reactions using only halogen compounds and more especially to a process the alkali metal fluorides as proposed hitherto. The fur for the production of highly fluorinated aromatic com ther possibility of using a continuous flow apparatus such pounds by the replacement of higher halogen atoms in 15 as a fluidised reactor will be apparent to those familiar halogeno-aromatic compounds by fluorine atoms. with the art. The presence of the two fluorides or com Aromatic halogenocarbons containing carbon and halo plex fluoride enables a lower temperature to be em gen atoms only can be reacted with alkali fluorides ployed than was hitherto believed to be necessary, with under various conditions to give yields of halofluoro a consequent reduction in the extent of thermal degrada aromatic compounds. -

Pacs by Chemical Name (Mg/M3) (Pdf)
Table 4: Protective Action Criteria (PAC) Rev 25 based on applicable 60-minute AEGLs, ERPGs, or TEELs. Values are presented in mg/m3. August 2009 Table 4 is an alphabetical listing of the chemicals in the PAC data set. It provides Chemical Abstract Service Registry Numbers (CASRNs)1, PAC values, and technical information on the source of the PAC values. Table 4 presents all values for TEEL-0, PAC-1, PAC-2, and PAC-3 in mg/m3. The conversion of ppm to mg/m3 is calculated assuming 25 ºC and 760 mm Hg. The columns presented in Table 4 provide the following information: Heading Definition No. The ordered numbering of the chemicals as they appear in this alphabetical listing. Chemical Name The chemical name given to the PAC Development Team. CASRN The Chemical Abstract Service Registry Number for this chemical. TEEL-0 This is the threshold concentration below which most people will experience no adverse health effects. This PAC is always based on TEEL-0 because AEGL-0 or ERPG-0 values do not exist. PAC-1 Based on the applicable AEGL-1, ERPG-1, or TEEL-1 value. PAC-2 Based on the applicable AEGL-2, ERPG-2, or TEEL-2 value. PAC-3 Based on the applicable AEGL-3, ERPG-3, or TEEL-3 value. Source of PACs: Technical comments provided by the PAC development team that TEEL-0, PAC-1, indicate the source of the data used to derive PAC values. Future efforts PAC-2, PAC-3 are directed at reviewing, revising, and enhancing this information. -
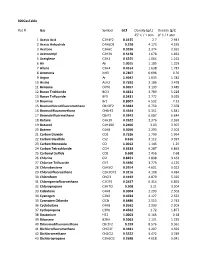
Gas Conversion Factor for 300 Series
300GasTable Rec # Gas Symbol GCF Density (g/L) Density (g/L) 25° C / 1 atm 0° C / 1 atm 1 Acetic Acid C2H4F2 0.4155 2.7 2.947 2 Acetic Anhydride C4H6O3 0.258 4.173 4.555 3 Acetone C3H6O 0.3556 2.374 2.591 4 Acetonitryl C2H3N 0.5178 1.678 1.832 5 Acetylene C2H2 0.6255 1.064 1.162 6 Air Air 1.0015 1.185 1.293 7 Allene C3H4 0.4514 1.638 1.787 8 Ammonia NH3 0.7807 0.696 0.76 9 Argon Ar 1.4047 1.633 1.782 10 Arsine AsH3 0.7592 3.186 3.478 11 Benzene C6H6 0.3057 3.193 3.485 12 Boron Trichloride BCl3 0.4421 4.789 5.228 13 Boron Triflouride BF3 0.5431 2.772 3.025 14 Bromine Br2 0.8007 6.532 7.13 15 Bromochlorodifluoromethane CBrClF2 0.3684 6.759 7.378 16 Bromodifluoromethane CHBrF2 0.4644 5.351 5.841 17 Bromotrifluormethane CBrF3 0.3943 6.087 6.644 18 Butane C4H10 0.2622 2.376 2.593 19 Butanol C4H10O 0.2406 3.03 3.307 20 Butene C4H8 0.3056 2.293 2.503 21 Carbon Dioxide CO2 0.7526 1.799 1.964 22 Carbon Disulfide CS2 0.616 3.112 3.397 23 Carbon Monoxide CO 1.0012 1.145 1.25 24 Carbon Tetrachloride CCl4 0.3333 6.287 6.863 25 Carbonyl Sulfide COS 0.668 2.456 2.68 26 Chlorine Cl2 0.8451 2.898 3.163 27 Chlorine Trifluoride ClF3 0.4496 3.779 4.125 28 Chlorobenzene C6H5Cl 0.2614 4.601 5.022 29 Chlorodifluoroethane C2H3ClF2 0.3216 4.108 4.484 30 Chloroform CHCl3 0.4192 4.879 5.326 31 Chloropentafluoroethane C2ClF5 0.2437 6.314 6.892 32 Chloropropane C3H7Cl 0.308 3.21 3.504 33 Cisbutene C4H8 0.3004 2.293 2.503 34 Cyanogen C2N2 0.4924 2.127 2.322 35 Cyanogen Chloride ClCN 0.6486 2.513 2.743 36 Cyclobutane C4H8 0.3562 2.293 2.503 37 Cyclopropane C3H6 0.4562 -

Synthesis of Hexafluorobenzene Through Batch Reactive Distillation
SYNTHESIS OF HEXAFLUOROBENZENE THROUGH BATCH REACTIVE DISTILLATION Jesita Reddy [Bsc. Eng.] In fulfilment of the requirements for the degree Master of Science in Engineering, College of Agriculture, Engineering and Science, University of KwaZulu-Natal December 2018 Supervisors: Doctor David Lokhat and Professor Deresh Ramjugernath As the candidate’s supervisor I agree to the submission of this thesis: Dr. D. Lokhat Prof. D. Ramjugernath DECLARATION I, Jesita Reddy declare that: The research reported in this dissertation/thesis, except where otherwise indicated, is my original work. 1. The research reported in this thesis, except where otherwise indicated, is my original research. 2. This thesis has not been submitted for any degree or examination at any other university. 3. This thesis does not contain other persons’ data, pictures, graphs or other information, unless specifically acknowledged as being sourced from other persons. 4. This thesis does not contain other persons' writing, unless specifically acknowledged as being sourced from other researchers. Where other written sources have been quoted, then: i. Their words have been re-written but the general information attributed to them has been referenced ii. Where their exact words have been used, then their writing has been placed in italics and inside quotation marks, and referenced. 5. This thesis does not contain text, graphics or tables copied and pasted from the Internet, unless specifically acknowledged, and the source being detailed in the thesis and in the References sections. Signed: ________________ Date: ____13/03/19____________ ii ACKNOWLEDGEMENTS This research was affiliated with the Fluorochemical Expansion Initiative recognized by the Department of Science and Technology. -

Tetramethylammonium Fluoride Tetrahydrate for Snar Fluorination of 4‑Chlorothiazoles at a Production Scale Mai Khanh Hawk,* Sarah J
pubs.acs.org/OPRD Article Tetramethylammonium Fluoride Tetrahydrate for SNAr Fluorination of 4‑Chlorothiazoles at a Production Scale Mai Khanh Hawk,* Sarah J. Ryan,* Xin Zhang, Ping Huang, Jing Chen, Chuanren Liu, Jianping Chen, Peter J. Lindsay-Scott, John Burnett, Craig White, Yu Lu, and John R. Rizzo Cite This: https://doi.org/10.1021/acs.oprd.1c00042 Read Online ACCESS Metrics & More Article Recommendations *sı Supporting Information fl · ABSTRACT: This article describes the use of tetramethylammonium uoride tetrahydrate (TMAF 4H2O) for the large-scale fl · preparation of a challenging 4- uorothiazole. Commercially available TMAF 4H2O was procured on a large scale and rigorously dried by distillation with isopropyl alcohol and then dimethylformamide at elevated temperature. This method of drying provided anhydrous TMAF [TMAF (anh)] containing <0.2 wt % water and <60 ppm isopropanol. The use of TMAF (anh) was essential for production of the 4-fluorothiazole. When the chlorothiazole starting material was treated with other anhydrous fluoride sources, fl · poor conversion of the starting material or potential safety issues were observed. SNAr uorination using dried TMAF 4H2O was carried out at a 45.1 kg scale at 95−100 °C to produce 36.8 kg of 4-fluorothiazole 1b. fl fl fl fl KEYWORDS: uorination, SNAr, tetramethylammonium uoride, uorothiazole, heteroarene, anhydrous uoride ■ INTRODUCTION demonstrated that anhydrous tetrabutylammonium fluoride fl [TBAF (anh)] could be prepared in situ from tetrabutylam- The substitution of hydrogen for uorine can result in fl monium cyanide (TBACN) and hexa uorobenzene (C6F6) improved bioavailability or metabolic stability of bioactive 11 1 (Scheme 2A). This reagent was used for the room- molecules. -

20210311 IAEG AD-DSL V5.0 for Pdf.Xlsx
IAEGTM AD-DSL Release Version 4.1 12-30-2020 Authority: IAEG Identity: AD-DSL Version number: 4.1 Issue Date: 2020-12-30 Key Yellow shading indicates AD-DSL family group entries, which can be expanded to display a non-exhaustive list of secondary CAS numbers belonging to the family group Substance Identification Change Log IAEG Regulatory Date First Parent Group IAEG ID CAS EC Name Synonyms Revision Date ECHA ID Entry Type Criteria Added IAEG ID IAEG000001 1327-53-3 215-481-4 Diarsenic trioxide Arsenic trioxide R1;R2;D1 2015-03-17 2015-03-17 100.014.075 Substance Direct Entry IAEG000002 1303-28-2 215-116-9 Diarsenic pentaoxide Arsenic pentoxide; Arsenic oxide R1;R2;D1 2015-03-17 2015-03-17 100.013.743 Substance Direct Entry IAEG000003 15606-95-8 427-700-2 Triethyl arsenate R1;R2;D1 2015-03-17 2017-08-14 100.102.611 Substance Direct Entry IAEG000004 7778-39-4 231-901-9 Arsenic acid R1;R2;D1 2015-03-17 2015-03-17 100.029.001 Substance Direct Entry IAEG000005 3687-31-8 222-979-5 Trilead diarsenate R1;R2;D1 2015-03-17 2017-08-14 100.020.890 Substance Direct Entry IAEG000006 7778-44-1 231-904-5 Calcium arsenate R1;R2;D1 2015-03-17 2017-08-14 100.029.003 Substance Direct Entry IAEG000009 12006-15-4 234-484-1 Cadmium arsenide Tricadmium diarsenide R1;R2;D1 2017-08-14 2017-08-14 Substance Direct Entry IAEG000021 7440-41-7 231-150-7 Beryllium (Be) R2 2015-03-17 2019-01-24 Substance Direct Entry IAEG000022 1306-19-0 215-146-2 Cadmium oxide R1;R2;D1 2015-03-17 2017-08-14 100.013.770 Substance Direct Entry IAEG000023 10108-64-2 233-296-7 Cadmium -

United States Patent Office
3,251,390 United States Patent Office Patented May 17, 1966 2 That the pyrolysis reactions which are the subject of 3,251,890 PRODUCTION OF HEXAFLUOROBENZENE the present invention could be carried out was wholly Robert Neville Haszeldine, “Windyridge, Lyme Road, Surprising in that it was to be expected that pyrolysis of Disley, England, and John Michael Birchall, Flat 3, a fluoro- or fluorochloro olefin would yield either a 77 Palatine Road, Withington, Manchester 20, England cyclobutane, or an acyclic compound, likely a dimer, No Drawing. Filed Oct. 30, 1961, Ser. No. 149,818 trimer or tetramer. Thus, for example, it is well known Claims priority, application Great Britain, Nov. 17, 1959, that the pyrolysis of CF:CFC1 yields either the cyclo 38,961/59 butane 4 Claims. (C. 260-650) 0. This application is a continuation-in-part of our co CF-CFCQF-GFCI pending application Serial No. 37,842, filed June 22, 1960, now abandoned. or the dimer CFC1 CFC-CF: CF. Again, the fluoro This invention relates to a new chemical process and chloro olefin CFCFCFCI on pyrolysis gives a linear in particular to a new process for the preparation of dimer, trimer or tetramer. Yet again, perfluoroethylene hexafluorobenzene. when pyrolysed at 200 C. yields perfluorocyclobutane Hexafluorobenzene is of great interest in organic and at 500 C. gives perfluoroisobutylene and also per fluorine chemistry as it is analogous to benzene in class fluoropropene. ical aromatic organic chemistry. It is valuable as an in One Way of conducting the pyrolysis reaction is to pass termediate in the preparation of pharmaceutical com 20 a vapor stream of the starting compound through a heated pounds and of polymers, such as the polyethers derived tube in the absence of moisture, air or oxygen. -

ANHYDROUS FLUORIDE SALTS and REAGENTS and METHODS for THEIR PRODUCTION Stephen G
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Faculty Publications -- Chemistry Department Published Research - Department of Chemistry 2009 ANHYDROUS FLUORIDE SALTS AND REAGENTS AND METHODS FOR THEIR PRODUCTION Stephen G. DiMagno Lincoln, NE, [email protected] Haoran Sun Lincoln, NE Follow this and additional works at: https://digitalcommons.unl.edu/chemfacpub Part of the Analytical Chemistry Commons, Medicinal-Pharmaceutical Chemistry Commons, and the Other Chemistry Commons DiMagno, Stephen G. and Sun, Haoran, "ANHYDROUS FLUORIDE SALTS AND REAGENTS AND METHODS FOR THEIR PRODUCTION" (2009). Faculty Publications -- Chemistry Department. 185. https://digitalcommons.unl.edu/chemfacpub/185 This Article is brought to you for free and open access by the Published Research - Department of Chemistry at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Faculty Publications -- Chemistry Department by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. US007592486B2 (12) United States Patent (10) Patent No.: US 7,592.486 B2 DiMagno et al. (45) Date of Patent: Sep. 22, 2009 (54) ANHYDROUS FLUORIDE SALTS AND 6,794,401 B2 9/2004 Nag REAGENTS AND METHODS FOR THEIR PRODUCTION FOREIGN PATENT DOCUMENTS CA 2035561 11, 1991 (75) Inventors: Stephen G. DiMagno, Lincoln, NE OTHER PUBLICATIONS (US); Haoran Sun, Lincoln, NE (US) CheMBioChemSpecial Issue: Fluorine in the Life Sciences 2004, 5, (73) Assignee:rsr rr l Regent oths laterily of Christe,557726. K.O., et al., J. Am. Chem. Soc. 1990, 112, 7619-25. eDraska, L1ncoln, Gnann, R.Z., et al., J. Am. Chem. Soc. 1997, 119, 112-115. (*) Notice: Subject to any disclaimer, the term of this JyK th,1990, A., et al.y ...."I . -

Toxicological Profile for Hexachlorobenzene
HEXACHLOROBENZENE 317 9. REFERENCES Abd-Allah AMA. 1999. Organochlorine contaminants in microlayer and subsurface water of Alexandria Coast, Egypt. J AOAC Int 82(2):391-398. Abraham K, Hille A, Ende M, et al. 1994. Intake and fecal excretion of PCDDs, PCDFs, HCB and PCBs (138,153,180) in a breast-fed and a formula-fed infant. Chemosphere 29(9-11):2279-2286. Abraham K, Papke O, Wahn U, et al. 2000. POP accumulation in infants during breast feeding. Organohalogen Compounds 48:25-26. ACGIH. 2014. Hexachlorobenzene. In: TLVs and BEIs based on the documentation of the threshold limit values for chemical substances and physical agents and biological exposure indices. Cincinnati, OH: American Conference of Governmental Industrial Hygienists, 33, 72-77. Ackerman LK, Schwindt AR, Simonich SL, et al. 2008. Atmospherically deposited PBDEs, pesticides, PCBs, and PAHs in Western U.S. National Park fish: Concentrations and consumption guidelines. Environ Sci Technol 42(7):2334-2341. Addae C, Cheng H, Martinez-Ceballos E. 2013. Effect of the environmental pollutant hexachlorobenzene (HCB) on the neuronal differentiation of mouse embryonic stem cells. Int J Environ Res Public Health 10(10):5244-5256. Adjarov DG. 1990. Decreased activity of liver coproporphyrinogen oxidase in hexachlorobenzene induced porphyria. Exp Pathol 40:117-122. *Adjarov DJ, Elder GH. 1986. Accumulation of uroporphyrin does not provoke further inhibition of liver uroporphyrinogen decarboxylase activity in hexachlorobenzene-induced porphyria. In: Hexachlorobenzene: Proceedings of an international symposium. IARC Sci Publ 77:467-469. Adjarov D, Ivanov E, Keremidchiev D. 1982. Gamma-glutamyl transferase: A sensitive marker in experimental hexachlorobenzene intoxication. Toxicology 23:73-77. -

Gamma Irradiation of Hexafluorobenzene1 R
JOURNAL OF RESEARCH of the National Bureau of Standards-A. Physics and Chemi stry Vol. 64A, No. 4, July- August 1960 Gamma Irradiation of Hexafluorobenzene1 R. E. Florin, 1. A. Wall, and D. W. Brown r, (March 27, 1960) Mi xtures of hexafluorobenze ne a,nd benzene were irradi ated in li quid phase by means of a C060 cram ma ource at 20° a nd a t '2 18° C. P crfiuorohep tane a nd vari ous binar.v mixt ures involvi~ g perfluorohepta nc, hexaflu or obe ll ze ne, be nze ne, and cyclo hexane were a lso in'adi ated at 20° C. H exafluorobenzene rese mbled benzene very closely in its behavior upon r adio\ysis. Generall y t he fluorocarbon-hydrocarbon mixt ures evolved much mo rc SiF4 (indicatin g the formation of J-IF, which r eacts with the glass vessel) than the pure flu oro carbon components. The polymer from hexaflu o robenzene-benzrne mixtures was p r oba~ly rich in cyclohexadiene and cyclohexene un its, I' csembiing t hat from pu re benzene, a nd Its co mposition ratio exhibited a strong "altern atin g" tendency. The res ults a re discussed in terms of free-radical and excited-state mechanisms. r\ t 218 0 C hexaflu orobenzene a nd a lso its mixtures with benze ne showed qualitative difl'erences from their behavior a t 20° C, a ltho ugh t he G va lues for SiF4 and polymer rema ined moderate. 1. Introduction more resistant than hydrocarbons to heat, and also ftromatic compounds more resistant than aliphatic.