Endothelial Dysfunction in Atherosclerosis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Cardiopulmonary Effects of the Calcitonin Gene- Related Peptide Family Kalsitonin-Geni İle İlişkili Peptit Ailesinin Kardiyopulmoner Etkileri
Turk J Pharm Sci 2020;17(3):349-356 DOI: 10.4274/tjps.galenos.2019.47123 REVIEW The Cardiopulmonary Effects of the Calcitonin Gene- related Peptide Family Kalsitonin-Geni İle İlişkili Peptit Ailesinin Kardiyopulmoner Etkileri Gökçen TELLİ1, Banu Cahide TEL1, Bülent GÜMÜŞEL2* 1Hacettepe University Faculty of Pharmacy, Department of Pharmacology, Ankara, Turkey 2Lokman Hekim University Faculty of Pharmacy, Department of Pharmacology, Ankara, Turkey ABSTRACT Cardiopulmonary diseases are very common among the population. They are high-cost diseases and there are still no definitive treatments. The roles of members of the calcitonin-gene related-peptide (CGRP) family in treating cardiopulmonary diseases have been studied for many years and promising results obtained. Especially in recent years, two important members of the family, adrenomedullin and adrenomedullin2/intermedin, have been considered new treatment targets in cardiopulmonary diseases. In this review, the roles of CGRP family members in cardiopulmonary diseases are investigated based on the studies performed to date. Key words: CGRP family, cardiopulmonary diseases, adrenomedullin, adrenomedullin2/intermedin, pulmonary hypertension ÖZ Kardiyopulmoner hastalıklar toplumda sık görülen, tedavi maliyeti oldukça yüksek ve halen kesin bir tedavisi bulunmayan hastalıklardır. Kalsitonin- geni ile ilişkili peptit (CGRP) ailesinin üyelerinin bir çok kardiyopulmoner hastalıktaki rolleri uzun yıllardır çalışılmakta ve umut vadeden sonuçlar elde edilmektedir. Özellikle son yıllarda CGRP ailesine -
Supporting Information
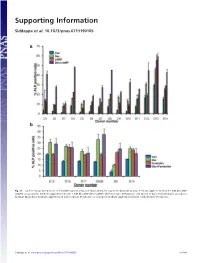
Supporting Information Siddappa et al. 10.1073/pnas.0711190105 Fig. S1. (a) Percentage ALP-positive cells in hMSCs grown in basic medium (Con), osteogenic medium (Dex), basic medium supplemented with 1 mM db-cAMP (cAMP), or osteogenic medium supplemented with 1 mM db-cAMP (DexϩcAMP). (b) Percentage ALP-positive cells grown in basic medium (Con), osteogenic medium (Dex), basic medium supplemented with forskolin (Forskolin), or osteogenic medium supplemented with forskolin (DexϩForskolin). Siddappa et al. www.pnas.org/cgi/content/short/0711190105 1of10 Fig. S2. hMSCs were grown in basic medium, basic medium supplemented with 1 mM db-cAMP (cAMP), osteogenic medium (Dex), or osteogenic medium supplemented with 1 mM db-cAMP (DexϩcAMP). Expression was analyzed by qPCR and is expressed as fold induction compared with cells grown in basic medium. The data were analyzed by using two-way ANOVA, and statistical significance is indicated compared with cells grown in basic medium. *, P Ͻ 0.05. Siddappa et al. www.pnas.org/cgi/content/short/0711190105 2of10 Fig. S3. (a) Methylene blue staining of hMSC-seeded scaffolds grown in basic medium (Con) or basic medium supplemented with 1 mM db-cAMP (cAMP) for 4 days. Note the less intensely stained db-cAMP-treated construct, indicating reduced cell numbers. (b) Quantitative Alamar blue assay for cell number analysis. The data were analyzed by using one-way ANOVA followed by Dunnet’s multiple-comparison test. Statistical significance is indicated compared with cells grown in basic medium (Con). *, P Ͻ 0.05 Siddappa et al. www.pnas.org/cgi/content/short/0711190105 3of10 Fig. -

Table of Contents
Therapeutic systems for Insulin-like growth factor-I Dissertation zur Erlangung des naturwissenschaftlichen Doktorgrades der Julius-Maximilians-Universität Würzburg vorgelegt von Isabel Schultz aus Dahn Würzburg 2015 Eingereicht bei der Fakultät für Chemie und Pharmazie am Gutachter der schriftlichen Arbeit 1. Gutachter: 2. Gutachter: Prüfer des öffentlichen Promotionskolloquiums 1. Prüfer: 2. Prüfer: 3. Prüfer: Datum des öffentlichen Promotionskolloquiums Doktorurkunde ausgehändigt am TABLE OF CONTENTS TABLE OF CONTENTS SUMMARY ............................................................................... 1 ZUSAMMEMFASSUNG .......................................................... 5 CHAPTER I ............................................................................... 9 DRUG DELIVERY OF INSULIN-LIKE GROWTH FACTOR I CHAPTER II ............................................................................ 45 INSULIN-LIKE GROWTH FACTOR-I AEROSOL FORMULATIONS FOR PULMONARY DELIVERY CHAPTER III ........................................................................... 73 PULMONARY INSULIN-LIKE GROWTH FACTOR I DELIVERY FROM TREHALOSE AND SILK-FIBROIN MICROPARTICLES CHAPTER IV ......................................................................... 113 EXPRESSION OF IGF-I MUTANTS CONCLUSION AND OUTLOOK ......................................... 147 DOCUMENTATION OF AUTHORSHIP ............................. 159 CURRICULUM VITAE ......................................................... 163 ACKNOWLEDGMENTS ..................................................... -

Thymic Mesenchymal Cells Have a Distinct Transcriptomic Profile
Thymic Mesenchymal Cells Have a Distinct Transcriptomic Profile Julien Patenaude and Claude Perreault This information is current as J Immunol 2016; 196:4760-4770; Prepublished online 29 of October 1, 2021. April 2016; doi: 10.4049/jimmunol.1502499 http://www.jimmunol.org/content/196/11/4760 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2016/04/29/jimmunol.150249 Material 9.DCSupplemental References This article cites 65 articles, 18 of which you can access for free at: http://www.jimmunol.org/content/196/11/4760.full#ref-list-1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on October 1, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Thymic Mesenchymal Cells Have a Distinct Transcriptomic Profile Julien Patenaude and Claude Perreault In order to understand the role of mesenchymal cells (MCs) in the adult thymus, we performed whole transcriptome analyses of primary thymic, bone, and skin MCs. -

Growth Hormone and Insulin-Like Growth Factors I and II Produce
Proc. Natl. Acad. Sci. USA Vol. 82, pp. 8724-8728, December 1985 Medical Sciences Growth hormone and insulin-like growth factors I and II produce distinct alterations in glucose metabolism in 3T3-F442A adipocytes (glucose oxidation/lipid accumulation/somatomedin C/multiplication-stimulating activity/recombinant DNA-derived insulin-like growth factor I) JESSICA SCHWARTZ, CAROL M. FOSTER, AND MARTA S. SATIN Department of Physiology, University of Michigan Medical School, Ann Arbor, MI 48109 Communicated by H. W. Davenport, August 20, 1985 ABSTRACT In 3T3-F442A adipocytes, human growth difficult (9, 10) because the onset of the suppressive effects hormone (hGH) stimulates glucose oxidation in 4 hr. A requires several days and in vitro preparations sensitive to maximal increase is evident at hGH concentrations of 50-100 GH (e.g., adipocytes) are not viable for extended periods. ng/ml and rarely exceeds 50% above control. The stimulation Recently, we reported that cultured 3T3-F442A adipocytes is transient; after 48 hr of incubation with GH, glucose could be used to study the metabolic effects of GH (11). The oxidation is significantly suppressed to 35% below control differentiation of 3T3-F442A fibroblasts to adipocytes has values. In view of the concept that insulin-like growth factors been shown to be GH-dependent (12), although GH is not (IGF) may mediate the effects of GH, we compared the effects required to maintain the adipocytes. We have found that, ofhGH (500 ng/ml) and several preparations ofIGF on glucose once converted, the 3T3 adipocytes are sensitive to both metabolism in 3T3 adipocytes. After 4 hr of incubation, IGF-I stimulatory and suppressive effects of human GH (hGH) on from human plasma stimulated glucose oxidation in a dose- glucose metabolism (11). -

Nitric Oxide As a Second Messenger in Parathyroid Hormone-Related Protein Signaling
433 Nitric oxide as a second messenger in parathyroid hormone-related protein signaling L Kalinowski, L W Dobrucki and T Malinski Department of Chemistry and Biochemistry, Ohio University, Athens, Ohio, USA (Requests for offprints should be addressed to T Malinski, Department of Chemistry and Biochemistry, Ohio University, Biochemistry Research Laboratories 136, Athens, Ohio 45701–2979, USA; Email: [email protected]) (L Kalinowski was on sabbatical leave from the Department of Clinical Biochemistry, Medical University of Gdansk and Laboratory of Cellular and Molecular Nephrology, Medical Research Center of the Polish Academy of Science, Poland) Abstract Parathyroid hormone (PTH)-related protein (PTHrP) is competitive PTH/PTHrP receptor antagonists, 10 µmol/l produced in smooth muscles and endothelial cells and [Leu11,-Trp12]-hPTHrP(7–34)amide and 10 µmol/l is believed to participate in the local regulation of vascu- [Nle8,18,Tyr34]-bPTH(3–34)amide, were equipotent in lar tone. No direct evidence for the activation of antagonizing hPTH(1–34)-stimulated NO release; endothelium-derived nitric oxide (NO) signaling pathway [Leu11,-Trp12]-hPTHrP(7–34)amide was more potent by PTHrP has been found despite attempts to identify it. than [Nle8,18,Tyr34]-bPTH(3–34)amide in inhibiting Based on direct in situ measurements, it is reported here for hPTHrP(1–34)-stimulated NO release. The PKC inhibi- the first time that the human PTH/PTHrP receptor tor, H-7 (50 µmol/l), did not change hPTH(1–34)- and analogs, hPTH(1–34) and hPTHrP(1–34), stimulate NO hPTHrP(1–34)-stimulated NO release, whereas the release from a single endothelial cell. -

Recent Advances in Endothelin Research on Cardiovascular and Endocrine Systems
Endocrine Journal 1994, 41(5), 491-507 Review Recent Advances in Endothelin Research on Cardiovascular and Endocrine Systems MITSUHIDE NARUSE, KIYoxo NARUSE, AND HIR0SH1 DEMURA Department of Medicine, Institute of Clinical Endocrinology, TokyoWomen's Medical College, Tokyo162, Japan Introduction Structure of ET and ET Receptor Since the quite exciting discovery in 1988 of The molecular and biochemical aspects of ET endothelin (ET) in the culture medium of aortic en- family peptides and ET receptors have been re- dothelial cells by Yanagisawa and coworkers [1], viewed by Masaki and Yanagisawa [13] and others evidence has accumulated to support its important [14-16]. We here describe the fundamentals. roles in the regulation of blood pressure and body fluid homeostasis and its pathophysiological sig- 1. ET Peptides nificance in various cardiovascular diseases. The discovery of ET and the opponent substances in- The ET molecule consists of 21 amino acid resi- cluding nitric oxide [2] and natriuretic peptides [3] dues with two intramolecular disulfide bonds be- during the last decade was epoch-making in the tween cystein residues at 1 and 15 and 3 and 11, field of "Cardiovascular Endocrinology". respectively. The ring structure and the hydropho- Because of its impressive potency and the long bic amino acid residues at the C-terminus are in- duration of its pressor action [1], the roles of ET in dispensable to its full biological activities. There the regulation of cardiovascular homeostasis have are three distinct isof orms of ET: ET-1, ET-2, and been extensively studied. However, the co-expres- ET-3 [13]. The difference in the amino acid se- sion or close localization of ET and its receptors in quence is seen in the ring structure, while they various tissues other than the vascular vessels sug- share the same sequence in the linear C-terminal gest non-vascular roles of ET [4-6]. -

Glucagon-Like Peptide-1 and Its Class BG Protein–Coupled Receptors
1521-0081/68/4/954–1013$25.00 http://dx.doi.org/10.1124/pr.115.011395 PHARMACOLOGICAL REVIEWS Pharmacol Rev 68:954–1013, October 2016 Copyright © 2016 by The Author(s) This is an open access article distributed under the CC BY-NC Attribution 4.0 International license. ASSOCIATE EDITOR: RICHARD DEQUAN YE Glucagon-Like Peptide-1 and Its Class B G Protein–Coupled Receptors: A Long March to Therapeutic Successes Chris de Graaf, Dan Donnelly, Denise Wootten, Jesper Lau, Patrick M. Sexton, Laurence J. Miller, Jung-Mo Ahn, Jiayu Liao, Madeleine M. Fletcher, Dehua Yang, Alastair J. H. Brown, Caihong Zhou, Jiejie Deng, and Ming-Wei Wang Division of Medicinal Chemistry, Faculty of Sciences, Vrije Universiteit Amsterdam, Amsterdam, The Netherlands (C.d.G.); School of Biomedical Sciences, University of Leeds, Leeds, United Kingdom (D.D.); Drug Discovery Biology Theme and Department of Pharmacology, Monash Institute of Pharmaceutical Sciences, Parkville, Victoria, Australia (D.W., P.M.S., M.M.F.); Protein and Peptide Chemistry, Global Research, Novo Nordisk A/S, Måløv, Denmark (J.La.); Department of Molecular Pharmacology and Experimental Therapeutics, Mayo Clinic, Scottsdale, Arizona (L.J.M.); Department of Chemistry and Biochemistry, University of Texas at Dallas, Richardson, Texas (J.-M.A.); Department of Bioengineering, Bourns College of Engineering, University of California at Riverside, Riverside, California (J.Li.); National Center for Drug Screening and CAS Key Laboratory of Receptor Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, China (D.Y., C.Z., J.D., M.-W.W.); Heptares Therapeutics, BioPark, Welwyn Garden City, United Kingdom (A.J.H.B.); and School of Pharmacy, Fudan University, Zhangjiang High-Tech Park, Shanghai, China (M.-W.W.) Downloaded from Abstract. -

An Investig Atio N of the Anabolic
AN INVESTIGATION OF THE ANABOLIC ACTIONS OF BIOSYNTHETIC HUMAN GROWTH HORMONE AFTER INJURY BY BURNING A thesis sumitted for the degree of MASTER OF SURGERY in the UNIVERSITY OF LONDON H.J.C.R. Belcher, MB, BS(Lond), FRCS(Eng). The Blond-Mclndoe Centre, Queen Victoria Hospital, Holtye Rd, East Grinstead, SUSSEX. - 1 - ProQuest Number: U053257 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a com plete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest ProQuest U053257 Published by ProQuest LLC(2017). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States C ode Microform Edition © ProQuest LLC. ProQuest LLC. 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106- 1346 To my wife, Georgina - 2 - ABSTRACT Previous clinical trials in normal subjects and post-operative patients have shown that biosynthetic growth hormone preparations increase nitrogen retention. It has been suggested that their administration to injured patients may be beneficial. A clinical trial is presented of twelve adult burned patients of whom six were allocated to receive biosynthetic human growth hormone (somatropin) and six to form a control group. Injury by burning is followed by increases in resting energy expenditure and urinary nitrogen excretion, accompanied by insulin resistance and glucose intolerance. There is a generalised fall in plasma protein concentrations, including the somatomedin, insulin-like growth factor-I. -

Vascular Effects of Parathyroid Hormone and Parathyroid Hormone-Related Protein in the Split Hydronephrotic Rat Kidney
2827 Journal of Physiology (1995), 483.2, pp. 481-490 481 Vascular effects of parathyroid hormone and parathyroid hormone-related protein in the split hydronephrotic rat kidney K. Endlich *, T. Massfelder t*, J. J. Helwig t and M. Steinhausen t I. Physiologisches Institut der Universitat Heidelberg, D-69120 Heidelberg, Germany and tLaboratoire de Physiologie Cellulaire Re'nale, Universite Louis Pasteur, Strasbourg, France 1. The effects of locally applied parathyroid hormone-related protein (PTHRP), a putative autocrine/paracrine hormone, on vascular diameters and glomerular blood flow (GBF) in the split hydronephrotic rat kidney were studied. As PTHRP interacts with parathyroid hormone (PTH) receptors in all tissues tested so far, the effects of PTHRP were compared with those of PTH. 2. Preglomerular vessels dilated in a concentration- and time-dependent manner that was almost identical for PTH and PTHRP. A significant preglomerular vasodilatation (5-17%) occurred at a threshold concentration of 10-10 mol 1-1 PTH or PTHRP, which raised GBF by 20 + 2 and 31 + 4%, respectively (means + S.E.M., n = 6). PTH or PTHRP (10-7 mol 1-') increased preglomerular diameters (11-36%) and GBF (60 + 10 and 70 + 8%, respectively) to near maximum. The most prominent dilatation was located at the interlobular artery and at the proximal afferent arteriole. 3. Efferent arterioles were not affected by either PTH or PTHRP. 4. Estimated concentrations of half-maximal response (EC50) for preglomerular vasodilatation and GBF increase were in the nanomolar to subnanomolar range. 5. After inhibition of angiotensin I-converting enzyme by 2 x 10-6 mol kg-' quinapril i.v. -

Growth Hormone, IGF-1, and Metabolism
www.currentmedicalliterature.com VOLUME 2 | NUMBER 3 | 2009 Growth Growth Hormone, IGF-1, and Metabolism Supported by an unrestricted educational grant from This activity is jointly sponsored by the University of Kentucky College of Medicine and Remedica Medical Education and Publishing. The University of Kentucky is an equal opportunity university. VOLUME 2 | NUMBER 3 | 2009 Growth: Growth Hormone, IGF-1, and Metabolism EDITOR-IN-CHIEF CLINICAL EDITORS NORMAN LAVIN, MD, PhD, FACE, FAAP CRAIG ALTER, MD University of California Los Angeles, The Children’s Hospital of Philadelphia, Los Angeles, CA, USA Philadelphia, PA, USA ADVISORY Board CHARLES BUCHANAN, BSc, MB (Hons), MRCP, FRCPCH DEREK LEROITH, MD, PhD King’s College Hospital, London, UK Mount Sinai School of Medicine, New York, NY, USA HIRALAL MAHESHWARI, MD, PhD Northwestern University Medical School, EDWARD REITER, MD Chicago, IL, USA Tufts University School of Medicine, Springfield, MA, USA RICHARD ROSS, MD, FRCP University of Sheffield, Sheffield, UK Visit CML – Growth online at www.currentmedicalliterature.com The website provides access to new and archived content, a personalized CML Compass search engine, a “Paper of the Month” service, and more! RT107_3_CML_GrowthH_2_3_07.indd 1 22/4/09 10:56:57 CUrrent Medical literatUre JOUrnalS Current Medical Literature journals are designed to solve the problem of information overload for specialist physicians. Each journal is compiled by an editorial team of clinicians from an ongoing review of the international literature, and articles are selected for citation and review on the basis of their relevance to clinical practice. Other titles in the series include the following. For additional information on any of these titles, please contact us at the address below. -

Autocrine Endothelin-3/Endothelin Receptor B Signaling Maintains Cellular and Molecular Properties of Glioblastoma Stem Cells
Published OnlineFirst October 19, 2011; DOI: 10.1158/1541-7786.MCR-10-0563 Molecular Cancer Cancer Genes and Genomics Research Autocrine Endothelin-3/Endothelin Receptor B Signaling Maintains Cellular and Molecular Properties of Glioblastoma Stem Cells Yue Liu1, Fei Ye1,9, Kazunari Yamada1, Jonathan L. Tso1, Yibei Zhang1, David H. Nguyen1, Qinghua Dong1,10, Horacio Soto2, Jinny Choe1, Anna Dembo1, Hayley Wheeler1, Ascia Eskin3, Ingrid Schmid4, William H. Yong5,8, Paul S. Mischel5,8, Timothy F. Cloughesy6,8, Harley I. Kornblum7,8, Stanley F. Nelson3,8, Linda M. Liau2,8, and Cho-Lea Tso1,8 Abstract Glioblastoma stem cells (GSC) express both radial glial cell and neural crest cell (NCC)-associated genes. We report that endothelin 3 (EDN3), an essential mitogen for NCC development and migration, is highly produced by GSCs. Serum-induced proliferative differentiation rapidly decreased EDN3 production and downregulated the expression of stemness-associated genes, and reciprocally, two glioblastoma markers, EDN1 and YKL-40 transcripts, were induced. Correspondingly, patient glioblastoma tissues express low levels of EDN3 mRNA and high levels of EDN1 and YKL-40 mRNA. Blocking EDN3/EDN receptor B (EDNRB) signaling by an EDNRB antagonist (BQ788), or EDN3 RNA interference (siRNA), leads to cell apoptosis and functional impairment of tumor sphere formation and cell spreading/migration in culture and loss of tumorigenic capacity in animals. Using exogenous EDN3 as the sole mitogen in culture does not support GSC propagation, but it can rescue GSCs from undergoing cell apoptosis. Molecular analysis by gene expression profiling revealed that most genes downregulated by EDN3/EDNRB blockade were those involved in cytoskeleton organization, pause of growth and differentiation, and DNA damage response, implicating the involvement of EDN3/EDNRB signaling in maintaining GSC migration, undifferentiation, and survival.