Table of Contents
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

List of Approved Ndas for Biological Products That Were Deemed to Be Blas on March 23, 2020
List of Approved NDAs for Biological Products That Were Deemed to be BLAs on March 23, 2020 On March 23, 2020, an approved application for a biological product under section 505 of the Federal Food, Drug, and Cosmetic Act (FD&C Act) was deemed to be a license for the biological product under section 351 of the Public Health Service Act (PHS Act) (see section 7002(e)(4)(A) of the Biologics Price Competition and Innovation Act of 2009). To enhance transparency and facilitate planning for the March 23, 2020, transition date, FDA compiled a preliminary list of approved applications for biological products under the FD&C Act that were listed in FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations (the Orange Book) and that would be affected by this transition provision. FDA posted this list on the FDA website in December 2018, and periodically updated this list before the March 23, 2020, transition date. The September 2019 update to this preliminary list added certain administratively closed applications related to approved applications for biological products that were on the December 2018 version of this list. The January 2020 update to the preliminary list reflected a change to the definition of “biological product” made by the Further Consolidated Appropriations Act, 2020, which was enacted on December 20, 2019. Section 605 of this Act further amended the definition of a “biological product” in section 351(i) of the PHS Act to remove the parenthetical “(except any chemically synthesized polypeptide)” from the statutory category of “protein.” FDA has provided below a list of each approved application for a biological product under the FD&C Act that was deemed to be a license (i.e., an approved biologics license application (BLA)) for the biological product on March 23, 2020. -

View Annual Report
INSMED INC FORM 10-K (Annual Report) Filed 03/16/10 for the Period Ending 12/31/09 Address 8720 STONY POINT PARKWAY SUITE 200 RICHMOND, VA 23235 Telephone 804-565-3000 CIK 0001104506 Symbol INSM SIC Code 2834 - Pharmaceutical Preparations Industry Biotechnology & Drugs Sector Healthcare Fiscal Year 12/31 http://www.edgar-online.com © Copyright 2010, EDGAR Online, Inc. All Rights Reserved. Distribution and use of this document restricted under EDGAR Online, Inc. Terms of Use. UNITED STATES SECURITIES AND EXCHANGE COMMISSION Washington, D.C. 20549 FORM 10-K (Mark One) ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 For the fiscal year ended: December 31, 2009 OR TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 For the transition period from __________ to __________ Commission File Number 0-30739 INSMED INCORPORATED (Exact name of registrant as specified in its charter) Virginia 54 -1972729 (State or other jurisdiction of incorporation or organization) (I.R.S. employer identification no.) 8720 Stony Point Parkway Richmond, Virginia 23235 (804) 565 -3000 (Address of principal executive offices) (Registrant ’s telephone number including area code) Securities registered pursuant to Section 12(b) of the Act: Title of each class Name of each exchange on which registered Common Stock, par value $0.01/share Nasdaq Capital Market Securities registered pursuant to Section 12(g) of the Act: None Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. -
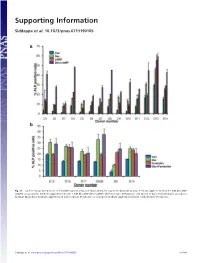
Supporting Information
Supporting Information Siddappa et al. 10.1073/pnas.0711190105 Fig. S1. (a) Percentage ALP-positive cells in hMSCs grown in basic medium (Con), osteogenic medium (Dex), basic medium supplemented with 1 mM db-cAMP (cAMP), or osteogenic medium supplemented with 1 mM db-cAMP (DexϩcAMP). (b) Percentage ALP-positive cells grown in basic medium (Con), osteogenic medium (Dex), basic medium supplemented with forskolin (Forskolin), or osteogenic medium supplemented with forskolin (DexϩForskolin). Siddappa et al. www.pnas.org/cgi/content/short/0711190105 1of10 Fig. S2. hMSCs were grown in basic medium, basic medium supplemented with 1 mM db-cAMP (cAMP), osteogenic medium (Dex), or osteogenic medium supplemented with 1 mM db-cAMP (DexϩcAMP). Expression was analyzed by qPCR and is expressed as fold induction compared with cells grown in basic medium. The data were analyzed by using two-way ANOVA, and statistical significance is indicated compared with cells grown in basic medium. *, P Ͻ 0.05. Siddappa et al. www.pnas.org/cgi/content/short/0711190105 2of10 Fig. S3. (a) Methylene blue staining of hMSC-seeded scaffolds grown in basic medium (Con) or basic medium supplemented with 1 mM db-cAMP (cAMP) for 4 days. Note the less intensely stained db-cAMP-treated construct, indicating reduced cell numbers. (b) Quantitative Alamar blue assay for cell number analysis. The data were analyzed by using one-way ANOVA followed by Dunnet’s multiple-comparison test. Statistical significance is indicated compared with cells grown in basic medium (Con). *, P Ͻ 0.05 Siddappa et al. www.pnas.org/cgi/content/short/0711190105 3of10 Fig. -

Us Anti-Doping Agency
2019U.S. ANTI-DOPING AGENCY WALLET CARDEXAMPLES OF PROHIBITED AND PERMITTED SUBSTANCES AND METHODS Effective Jan. 1 – Dec. 31, 2019 CATEGORIES OF SUBSTANCES PROHIBITED AT ALL TIMES (IN AND OUT-OF-COMPETITION) • Non-Approved Substances: investigational drugs and pharmaceuticals with no approval by a governmental regulatory health authority for human therapeutic use. • Anabolic Agents: androstenediol, androstenedione, bolasterone, boldenone, clenbuterol, danazol, desoxymethyltestosterone (madol), dehydrochlormethyltestosterone (DHCMT), Prasterone (dehydroepiandrosterone, DHEA , Intrarosa) and its prohormones, drostanolone, epitestosterone, methasterone, methyl-1-testosterone, methyltestosterone (Covaryx, EEMT, Est Estrogens-methyltest DS, Methitest), nandrolone, oxandrolone, prostanozol, Selective Androgen Receptor Modulators (enobosarm, (ostarine, MK-2866), andarine, LGD-4033, RAD-140). stanozolol, testosterone and its metabolites or isomers (Androgel), THG, tibolone, trenbolone, zeranol, zilpaterol, and similar substances. • Beta-2 Agonists: All selective and non-selective beta-2 agonists, including all optical isomers, are prohibited. Most inhaled beta-2 agonists are prohibited, including arformoterol (Brovana), fenoterol, higenamine (norcoclaurine, Tinospora crispa), indacaterol (Arcapta), levalbuterol (Xopenex), metaproternol (Alupent), orciprenaline, olodaterol (Striverdi), pirbuterol (Maxair), terbutaline (Brethaire), vilanterol (Breo). The only exceptions are albuterol, formoterol, and salmeterol by a metered-dose inhaler when used -

Clinical Policy: Mecasermin (Increlex) Reference Number: ERX.SPA.209 Effective Date: 01.11.17 Last Review Date: 11.17 Revision Log
Clinical Policy: Mecasermin (Increlex) Reference Number: ERX.SPA.209 Effective Date: 01.11.17 Last Review Date: 11.17 Revision Log See Important Reminder at the end of this policy for important regulatory and legal information. Description Mecasermin (Increlex®) is an insulin growth factor-1 (IGF-1) analogue. FDA Approved Indication(s) Increlex is indicated for the treatment of growth failure in children with severe primary IGF-1 deficiency or with growth hormone (GH) gene deletion who have developed neutralizing antibodies to GH. Limitation(s) of use: Increlex is not a substitute to GH for approved GH indications. Policy/Criteria Provider must submit documentation (which may include office chart notes and lab results) supporting that member has met all approval criteria It is the policy of health plans affiliated with Envolve Pharmacy Solutions™ that Increlex is medically necessary when the following criteria are met: I. Initial Approval Criteria A. Severe Primary IGF-1 Deficiency (must meet all): 1. Diagnosis of IGF-1 deficiency growth failure and associated growth failure with one of the following (a or b): a. Severe primary IGF-1 deficiency as defined by all (i through iii): i. Height standard deviation score (SDS) ≤ –3.0; ii. Basal IGF-1 SDS ≤ –3.0; iii. Normal or elevated GH level; b. GH gene deletion with development of neutralizing antibodies to GH; 2. Prescribed by or in consultation with an endocrinologist; 3. Age ≥ 2 and <18 years; 4. At the time of request, member does not have closed epiphyses; 5. Dose does not exceed 0.12 mg/kg twice daily. -

Mecasermin in Insulin Receptor-Related Severe Insulin Resistance Syndromes: Case Report and Review of the Literature
International Journal of Molecular Sciences Review Mecasermin in Insulin Receptor-Related Severe Insulin Resistance Syndromes: Case Report and Review of the Literature Michaela Plamper, Bettina Gohlke, Felix Schreiner and Joachim Woelfle * Pediatric Endocrinology and Diabetology Division, Children’s Hospital, University of Bonn, Adenauerallee 119, 53113 Bonn, Germany; [email protected] (M.P.); [email protected] (B.G.); [email protected] (F.S.) * Correspondence: Joachim.Woelfl[email protected]; Tel.: +49-228-2873-3223; Fax: +49-228-2873-3472 Received: 8 March 2018; Accepted: 18 April 2018; Published: 24 April 2018 Abstract: Mutations in the insulin receptor (INSR) gene underlie rare severe INSR-related insulin resistance syndromes (SIR), including insulin resistance type A, Rabson–Mendenhall syndrome and Donohue syndrome (DS), with DS representing the most severe form of insulin resistance. Treatment of these cases is challenging, with the majority of DS patients dying within the first two years of life. rhIGF-I (mecasermin) has been reported to improve metabolic control and increase lifespan in DS patients. A case report and literature review were completed. We present a case involving a male patient with DS, harbouring a homozygous mutation in the INSR gene (c.591delC). Initial rhIGF-I application via BID (twice daily) injection was unsatisfactory, but continuous subcutaneous rhIGF-I infusion via an insulin pump improved weight development and diabetes control (HbA1c decreased from 10 to 7.6%). However, our patient died at 22 months of age during the course of a respiratory infection in in Libya. Currently available data in the literature comprising more than 30 treated patients worldwide seem to support a trial of rhIGF-I in SIR. -

Thymic Mesenchymal Cells Have a Distinct Transcriptomic Profile
Thymic Mesenchymal Cells Have a Distinct Transcriptomic Profile Julien Patenaude and Claude Perreault This information is current as J Immunol 2016; 196:4760-4770; Prepublished online 29 of October 1, 2021. April 2016; doi: 10.4049/jimmunol.1502499 http://www.jimmunol.org/content/196/11/4760 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2016/04/29/jimmunol.150249 Material 9.DCSupplemental References This article cites 65 articles, 18 of which you can access for free at: http://www.jimmunol.org/content/196/11/4760.full#ref-list-1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on October 1, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Thymic Mesenchymal Cells Have a Distinct Transcriptomic Profile Julien Patenaude and Claude Perreault In order to understand the role of mesenchymal cells (MCs) in the adult thymus, we performed whole transcriptome analyses of primary thymic, bone, and skin MCs. -

Growth Hormone and Insulin-Like Growth Factors I and II Produce
Proc. Natl. Acad. Sci. USA Vol. 82, pp. 8724-8728, December 1985 Medical Sciences Growth hormone and insulin-like growth factors I and II produce distinct alterations in glucose metabolism in 3T3-F442A adipocytes (glucose oxidation/lipid accumulation/somatomedin C/multiplication-stimulating activity/recombinant DNA-derived insulin-like growth factor I) JESSICA SCHWARTZ, CAROL M. FOSTER, AND MARTA S. SATIN Department of Physiology, University of Michigan Medical School, Ann Arbor, MI 48109 Communicated by H. W. Davenport, August 20, 1985 ABSTRACT In 3T3-F442A adipocytes, human growth difficult (9, 10) because the onset of the suppressive effects hormone (hGH) stimulates glucose oxidation in 4 hr. A requires several days and in vitro preparations sensitive to maximal increase is evident at hGH concentrations of 50-100 GH (e.g., adipocytes) are not viable for extended periods. ng/ml and rarely exceeds 50% above control. The stimulation Recently, we reported that cultured 3T3-F442A adipocytes is transient; after 48 hr of incubation with GH, glucose could be used to study the metabolic effects of GH (11). The oxidation is significantly suppressed to 35% below control differentiation of 3T3-F442A fibroblasts to adipocytes has values. In view of the concept that insulin-like growth factors been shown to be GH-dependent (12), although GH is not (IGF) may mediate the effects of GH, we compared the effects required to maintain the adipocytes. We have found that, ofhGH (500 ng/ml) and several preparations ofIGF on glucose once converted, the 3T3 adipocytes are sensitive to both metabolism in 3T3 adipocytes. After 4 hr of incubation, IGF-I stimulatory and suppressive effects of human GH (hGH) on from human plasma stimulated glucose oxidation in a dose- glucose metabolism (11). -

The Use of Stems in the Selection of International Nonproprietary Names (INN) for Pharmaceutical Substances
WHO/PSM/QSM/2006.3 The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances 2006 Programme on International Nonproprietary Names (INN) Quality Assurance and Safety: Medicines Medicines Policy and Standards The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances FORMER DOCUMENT NUMBER: WHO/PHARM S/NOM 15 © World Health Organization 2006 All rights reserved. Publications of the World Health Organization can be obtained from WHO Press, World Health Organization, 20 Avenue Appia, 1211 Geneva 27, Switzerland (tel.: +41 22 791 3264; fax: +41 22 791 4857; e-mail: [email protected]). Requests for permission to reproduce or translate WHO publications – whether for sale or for noncommercial distribution – should be addressed to WHO Press, at the above address (fax: +41 22 791 4806; e-mail: [email protected]). The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted lines on maps represent approximate border lines for which there may not yet be full agreement. The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors and omissions excepted, the names of proprietary products are distinguished by initial capital letters. -

Recent Advances in Endothelin Research on Cardiovascular and Endocrine Systems
Endocrine Journal 1994, 41(5), 491-507 Review Recent Advances in Endothelin Research on Cardiovascular and Endocrine Systems MITSUHIDE NARUSE, KIYoxo NARUSE, AND HIR0SH1 DEMURA Department of Medicine, Institute of Clinical Endocrinology, TokyoWomen's Medical College, Tokyo162, Japan Introduction Structure of ET and ET Receptor Since the quite exciting discovery in 1988 of The molecular and biochemical aspects of ET endothelin (ET) in the culture medium of aortic en- family peptides and ET receptors have been re- dothelial cells by Yanagisawa and coworkers [1], viewed by Masaki and Yanagisawa [13] and others evidence has accumulated to support its important [14-16]. We here describe the fundamentals. roles in the regulation of blood pressure and body fluid homeostasis and its pathophysiological sig- 1. ET Peptides nificance in various cardiovascular diseases. The discovery of ET and the opponent substances in- The ET molecule consists of 21 amino acid resi- cluding nitric oxide [2] and natriuretic peptides [3] dues with two intramolecular disulfide bonds be- during the last decade was epoch-making in the tween cystein residues at 1 and 15 and 3 and 11, field of "Cardiovascular Endocrinology". respectively. The ring structure and the hydropho- Because of its impressive potency and the long bic amino acid residues at the C-terminus are in- duration of its pressor action [1], the roles of ET in dispensable to its full biological activities. There the regulation of cardiovascular homeostasis have are three distinct isof orms of ET: ET-1, ET-2, and been extensively studied. However, the co-expres- ET-3 [13]. The difference in the amino acid se- sion or close localization of ET and its receptors in quence is seen in the ring structure, while they various tissues other than the vascular vessels sug- share the same sequence in the linear C-terminal gest non-vascular roles of ET [4-6]. -

2021 Formulary List of Covered Prescription Drugs
2021 Formulary List of covered prescription drugs This drug list applies to all Individual HMO products and the following Small Group HMO products: Sharp Platinum 90 Performance HMO, Sharp Platinum 90 Performance HMO AI-AN, Sharp Platinum 90 Premier HMO, Sharp Platinum 90 Premier HMO AI-AN, Sharp Gold 80 Performance HMO, Sharp Gold 80 Performance HMO AI-AN, Sharp Gold 80 Premier HMO, Sharp Gold 80 Premier HMO AI-AN, Sharp Silver 70 Performance HMO, Sharp Silver 70 Performance HMO AI-AN, Sharp Silver 70 Premier HMO, Sharp Silver 70 Premier HMO AI-AN, Sharp Silver 73 Performance HMO, Sharp Silver 73 Premier HMO, Sharp Silver 87 Performance HMO, Sharp Silver 87 Premier HMO, Sharp Silver 94 Performance HMO, Sharp Silver 94 Premier HMO, Sharp Bronze 60 Performance HMO, Sharp Bronze 60 Performance HMO AI-AN, Sharp Bronze 60 Premier HDHP HMO, Sharp Bronze 60 Premier HDHP HMO AI-AN, Sharp Minimum Coverage Performance HMO, Sharp $0 Cost Share Performance HMO AI-AN, Sharp $0 Cost Share Premier HMO AI-AN, Sharp Silver 70 Off Exchange Performance HMO, Sharp Silver 70 Off Exchange Premier HMO, Sharp Performance Platinum 90 HMO 0/15 + Child Dental, Sharp Premier Platinum 90 HMO 0/20 + Child Dental, Sharp Performance Gold 80 HMO 350 /25 + Child Dental, Sharp Premier Gold 80 HMO 250/35 + Child Dental, Sharp Performance Silver 70 HMO 2250/50 + Child Dental, Sharp Premier Silver 70 HMO 2250/55 + Child Dental, Sharp Premier Silver 70 HDHP HMO 2500/20% + Child Dental, Sharp Performance Bronze 60 HMO 6300/65 + Child Dental, Sharp Premier Bronze 60 HDHP HMO -

Type 1 Diabetes
Type 1 Diabetes Clinical Management of the Athlete Ian Gallen Editor Type 1 Diabetes Clinical Management of the Athlete Editor Ian Gallen Diabetes Centre Wycombe Hospital High Wycombe UK ISBN 978-0-85729-753-2 e-ISBN 978-0-85729-754-9 DOI 10.1007/978-0-85729-754-9 Springer London Dordrecht Heidelberg New York British Library Cataloguing in Publication Data A catalogue record for this book is available from the British Library Library of Congress Control Number: 2012934821 © Springer-Verlag London Limited 2012 Apart from any fair dealing for the purposes of research or private study, or criticism or review, as permitted under the Copyright, Designs and Patents Act 1988, this publication may only be reproduced, stored or transmitted, in any form or by any means, with the prior permission in writing of the publishers, or in the case of reprographic reproduction in accordance with the terms of licenses issued by the Copyright Licensing Agency. Enquiries concerning reproduction outside those terms should be sent to the publishers. The use of registered names, trademarks, etc., in this publication does not imply, even in the absence of a specifi c statement, that such names are exempt from the relevant laws and regulations and therefore free for general use. Product liability: The publisher can give no guarantee for information about drug dosage and application thereof contained in this book. In every individual case the respective user must check its accuracy by consulting other pharmaceutical literature. Printed on acid-free paper Springer is part of Springer Science+Business Media (www.springer.com) This book is dedicated to my parents Louis and Barbara for their lifelong love, encouragement and support, to my wife Susan for our happy life together and to our children Robert and Hannah who make my life meaningful.