Growth Hormone, IGF-1, and Metabolism
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Supporting Information
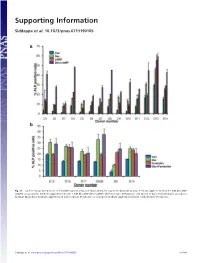
Supporting Information Siddappa et al. 10.1073/pnas.0711190105 Fig. S1. (a) Percentage ALP-positive cells in hMSCs grown in basic medium (Con), osteogenic medium (Dex), basic medium supplemented with 1 mM db-cAMP (cAMP), or osteogenic medium supplemented with 1 mM db-cAMP (DexϩcAMP). (b) Percentage ALP-positive cells grown in basic medium (Con), osteogenic medium (Dex), basic medium supplemented with forskolin (Forskolin), or osteogenic medium supplemented with forskolin (DexϩForskolin). Siddappa et al. www.pnas.org/cgi/content/short/0711190105 1of10 Fig. S2. hMSCs were grown in basic medium, basic medium supplemented with 1 mM db-cAMP (cAMP), osteogenic medium (Dex), or osteogenic medium supplemented with 1 mM db-cAMP (DexϩcAMP). Expression was analyzed by qPCR and is expressed as fold induction compared with cells grown in basic medium. The data were analyzed by using two-way ANOVA, and statistical significance is indicated compared with cells grown in basic medium. *, P Ͻ 0.05. Siddappa et al. www.pnas.org/cgi/content/short/0711190105 2of10 Fig. S3. (a) Methylene blue staining of hMSC-seeded scaffolds grown in basic medium (Con) or basic medium supplemented with 1 mM db-cAMP (cAMP) for 4 days. Note the less intensely stained db-cAMP-treated construct, indicating reduced cell numbers. (b) Quantitative Alamar blue assay for cell number analysis. The data were analyzed by using one-way ANOVA followed by Dunnet’s multiple-comparison test. Statistical significance is indicated compared with cells grown in basic medium (Con). *, P Ͻ 0.05 Siddappa et al. www.pnas.org/cgi/content/short/0711190105 3of10 Fig. -

Table of Contents
Therapeutic systems for Insulin-like growth factor-I Dissertation zur Erlangung des naturwissenschaftlichen Doktorgrades der Julius-Maximilians-Universität Würzburg vorgelegt von Isabel Schultz aus Dahn Würzburg 2015 Eingereicht bei der Fakultät für Chemie und Pharmazie am Gutachter der schriftlichen Arbeit 1. Gutachter: 2. Gutachter: Prüfer des öffentlichen Promotionskolloquiums 1. Prüfer: 2. Prüfer: 3. Prüfer: Datum des öffentlichen Promotionskolloquiums Doktorurkunde ausgehändigt am TABLE OF CONTENTS TABLE OF CONTENTS SUMMARY ............................................................................... 1 ZUSAMMEMFASSUNG .......................................................... 5 CHAPTER I ............................................................................... 9 DRUG DELIVERY OF INSULIN-LIKE GROWTH FACTOR I CHAPTER II ............................................................................ 45 INSULIN-LIKE GROWTH FACTOR-I AEROSOL FORMULATIONS FOR PULMONARY DELIVERY CHAPTER III ........................................................................... 73 PULMONARY INSULIN-LIKE GROWTH FACTOR I DELIVERY FROM TREHALOSE AND SILK-FIBROIN MICROPARTICLES CHAPTER IV ......................................................................... 113 EXPRESSION OF IGF-I MUTANTS CONCLUSION AND OUTLOOK ......................................... 147 DOCUMENTATION OF AUTHORSHIP ............................. 159 CURRICULUM VITAE ......................................................... 163 ACKNOWLEDGMENTS ..................................................... -

Thymic Mesenchymal Cells Have a Distinct Transcriptomic Profile
Thymic Mesenchymal Cells Have a Distinct Transcriptomic Profile Julien Patenaude and Claude Perreault This information is current as J Immunol 2016; 196:4760-4770; Prepublished online 29 of October 1, 2021. April 2016; doi: 10.4049/jimmunol.1502499 http://www.jimmunol.org/content/196/11/4760 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2016/04/29/jimmunol.150249 Material 9.DCSupplemental References This article cites 65 articles, 18 of which you can access for free at: http://www.jimmunol.org/content/196/11/4760.full#ref-list-1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on October 1, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Thymic Mesenchymal Cells Have a Distinct Transcriptomic Profile Julien Patenaude and Claude Perreault In order to understand the role of mesenchymal cells (MCs) in the adult thymus, we performed whole transcriptome analyses of primary thymic, bone, and skin MCs. -

Growth Hormone and Insulin-Like Growth Factors I and II Produce
Proc. Natl. Acad. Sci. USA Vol. 82, pp. 8724-8728, December 1985 Medical Sciences Growth hormone and insulin-like growth factors I and II produce distinct alterations in glucose metabolism in 3T3-F442A adipocytes (glucose oxidation/lipid accumulation/somatomedin C/multiplication-stimulating activity/recombinant DNA-derived insulin-like growth factor I) JESSICA SCHWARTZ, CAROL M. FOSTER, AND MARTA S. SATIN Department of Physiology, University of Michigan Medical School, Ann Arbor, MI 48109 Communicated by H. W. Davenport, August 20, 1985 ABSTRACT In 3T3-F442A adipocytes, human growth difficult (9, 10) because the onset of the suppressive effects hormone (hGH) stimulates glucose oxidation in 4 hr. A requires several days and in vitro preparations sensitive to maximal increase is evident at hGH concentrations of 50-100 GH (e.g., adipocytes) are not viable for extended periods. ng/ml and rarely exceeds 50% above control. The stimulation Recently, we reported that cultured 3T3-F442A adipocytes is transient; after 48 hr of incubation with GH, glucose could be used to study the metabolic effects of GH (11). The oxidation is significantly suppressed to 35% below control differentiation of 3T3-F442A fibroblasts to adipocytes has values. In view of the concept that insulin-like growth factors been shown to be GH-dependent (12), although GH is not (IGF) may mediate the effects of GH, we compared the effects required to maintain the adipocytes. We have found that, ofhGH (500 ng/ml) and several preparations ofIGF on glucose once converted, the 3T3 adipocytes are sensitive to both metabolism in 3T3 adipocytes. After 4 hr of incubation, IGF-I stimulatory and suppressive effects of human GH (hGH) on from human plasma stimulated glucose oxidation in a dose- glucose metabolism (11). -

Recent Advances in Endothelin Research on Cardiovascular and Endocrine Systems
Endocrine Journal 1994, 41(5), 491-507 Review Recent Advances in Endothelin Research on Cardiovascular and Endocrine Systems MITSUHIDE NARUSE, KIYoxo NARUSE, AND HIR0SH1 DEMURA Department of Medicine, Institute of Clinical Endocrinology, TokyoWomen's Medical College, Tokyo162, Japan Introduction Structure of ET and ET Receptor Since the quite exciting discovery in 1988 of The molecular and biochemical aspects of ET endothelin (ET) in the culture medium of aortic en- family peptides and ET receptors have been re- dothelial cells by Yanagisawa and coworkers [1], viewed by Masaki and Yanagisawa [13] and others evidence has accumulated to support its important [14-16]. We here describe the fundamentals. roles in the regulation of blood pressure and body fluid homeostasis and its pathophysiological sig- 1. ET Peptides nificance in various cardiovascular diseases. The discovery of ET and the opponent substances in- The ET molecule consists of 21 amino acid resi- cluding nitric oxide [2] and natriuretic peptides [3] dues with two intramolecular disulfide bonds be- during the last decade was epoch-making in the tween cystein residues at 1 and 15 and 3 and 11, field of "Cardiovascular Endocrinology". respectively. The ring structure and the hydropho- Because of its impressive potency and the long bic amino acid residues at the C-terminus are in- duration of its pressor action [1], the roles of ET in dispensable to its full biological activities. There the regulation of cardiovascular homeostasis have are three distinct isof orms of ET: ET-1, ET-2, and been extensively studied. However, the co-expres- ET-3 [13]. The difference in the amino acid se- sion or close localization of ET and its receptors in quence is seen in the ring structure, while they various tissues other than the vascular vessels sug- share the same sequence in the linear C-terminal gest non-vascular roles of ET [4-6]. -

An Investig Atio N of the Anabolic
AN INVESTIGATION OF THE ANABOLIC ACTIONS OF BIOSYNTHETIC HUMAN GROWTH HORMONE AFTER INJURY BY BURNING A thesis sumitted for the degree of MASTER OF SURGERY in the UNIVERSITY OF LONDON H.J.C.R. Belcher, MB, BS(Lond), FRCS(Eng). The Blond-Mclndoe Centre, Queen Victoria Hospital, Holtye Rd, East Grinstead, SUSSEX. - 1 - ProQuest Number: U053257 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a com plete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest ProQuest U053257 Published by ProQuest LLC(2017). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States C ode Microform Edition © ProQuest LLC. ProQuest LLC. 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106- 1346 To my wife, Georgina - 2 - ABSTRACT Previous clinical trials in normal subjects and post-operative patients have shown that biosynthetic growth hormone preparations increase nitrogen retention. It has been suggested that their administration to injured patients may be beneficial. A clinical trial is presented of twelve adult burned patients of whom six were allocated to receive biosynthetic human growth hormone (somatropin) and six to form a control group. Injury by burning is followed by increases in resting energy expenditure and urinary nitrogen excretion, accompanied by insulin resistance and glucose intolerance. There is a generalised fall in plasma protein concentrations, including the somatomedin, insulin-like growth factor-I. -

Supplemental Table 1
Supplemental Data Supplemental Table 1. Genes differentially regulated by Ad-KLF2 vs. Ad-GFP infected EC. Three independent genome-wide transcriptional profiling experiments were performed, and significantly regulated genes were identified. Color-coding scheme: Up, p < 1e-15 Up, 1e-15 < p < 5e-10 Up, 5e-10 < p < 5e-5 Up, 5e-5 < p <.05 Down, p < 1e-15 As determined by Zpool Down, 1e-15 < p < 5e-10 Down, 5e-10 < p < 5e-5 Down, 5e-5 < p <.05 p<.05 as determined by Iterative Standard Deviation Algorithm as described in Supplemental Methods Ratio RefSeq Number Gene Name 1,058.52 KRT13 - keratin 13 565.72 NM_007117.1 TRH - thyrotropin-releasing hormone 244.04 NM_001878.2 CRABP2 - cellular retinoic acid binding protein 2 118.90 NM_013279.1 C11orf9 - chromosome 11 open reading frame 9 109.68 NM_000517.3 HBA2;HBA1 - hemoglobin, alpha 2;hemoglobin, alpha 1 102.04 NM_001823.3 CKB - creatine kinase, brain 96.23 LYNX1 95.53 NM_002514.2 NOV - nephroblastoma overexpressed gene 75.82 CeleraFN113625 FLJ45224;PTGDS - FLJ45224 protein;prostaglandin D2 synthase 21kDa 74.73 NM_000954.5 (brain) 68.53 NM_205545.1 UNQ430 - RGTR430 66.89 NM_005980.2 S100P - S100 calcium binding protein P 64.39 NM_153370.1 PI16 - protease inhibitor 16 58.24 NM_031918.1 KLF16 - Kruppel-like factor 16 46.45 NM_024409.1 NPPC - natriuretic peptide precursor C 45.48 NM_032470.2 TNXB - tenascin XB 34.92 NM_001264.2 CDSN - corneodesmosin 33.86 NM_017671.3 C20orf42 - chromosome 20 open reading frame 42 33.76 NM_024829.3 FLJ22662 - hypothetical protein FLJ22662 32.10 NM_003283.3 TNNT1 - troponin T1, skeletal, slow LOC388888 (LOC388888), mRNA according to UniGene - potential 31.45 AK095686.1 CONFLICT - LOC388888 (na) according to LocusLink. -

Endocrine Test Selection and Interpretation
The Quest Diagnostics Manual Endocrinology Test Selection and Interpretation Fourth Edition The Quest Diagnostics Manual Endocrinology Test Selection and Interpretation Fourth Edition Edited by: Delbert A. Fisher, MD Senior Science Officer Quest Diagnostics Nichols Institute Professor Emeritus, Pediatrics and Medicine UCLA School of Medicine Consulting Editors: Wael Salameh, MD, FACP Medical Director, Endocrinology/Metabolism Quest Diagnostics Nichols Institute San Juan Capistrano, CA Associate Clinical Professor of Medicine, David Geffen School of Medicine at UCLA Richard W. Furlanetto, MD, PhD Medical Director, Endocrinology/Metabolism Quest Diagnostics Nichols Institute Chantilly, VA ©2007 Quest Diagnostics Incorporated. All rights reserved. Fourth Edition Printed in the United States of America Quest, Quest Diagnostics, the associated logo, Nichols Institute, and all associated Quest Diagnostics marks are the trademarks of Quest Diagnostics. All third party marks − ®' and ™' − are the property of their respective owners. No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopy, recording, and information storage and retrieval system, without permission in writing from the publisher. Address inquiries to the Medical Information Department, Quest Diagnostics Nichols Institute, 33608 Ortega Highway, San Juan Capistrano, CA 92690-6130. Previous editions copyrighted in 1996, 1998, and 2004. Re-order # IG1984 Forward Quest Diagnostics Nichols Institute has been -

Retinal and Choroidal Angiogenesis: a Review of New Targets Thiago Cabral1,2,3, Luiz Guilherme M
Cabral et al. Int J Retin Vitr (2017) 3:31 DOI 10.1186/s40942-017-0084-9 International Journal of Retina and Vitreous REVIEW Open Access Retinal and choroidal angiogenesis: a review of new targets Thiago Cabral1,2,3, Luiz Guilherme M. Mello3, Luiz H. Lima2, Júlia Polido2,3, Caio V. Regatieri2, Rubens Belfort Jr.2 and Vinit B. Mahajan4,5* Abstract Retinal and choroidal neovascularization are a major cause of signifcant visual impairment, worldwide. Understand- ing the various factors involved in the accompanying physiopathology is vital for development of novel treatments, and most important, for preserving patient vision. The intraocular use of anti-vascular endothelial growth factor thera- peutics has improved management of the retinal and choroidal neovascularization but some patients do not respond, suggesting other vascular mediators may also contribute to ocular angiogenesis. Several recent studies examined possible new targets for future anti-angiogenic therapies. Potential targets of retinal and choroidal neovascularization therapy include members of the platelet-derived growth factor family, vascular endothelial growth factor sub-family, epidermal growth factor family, fbroblast growth factor family, transforming growth factor-β superfamily (TGF-β1, activins, follistatin and bone morphogenetic proteins), angiopoietin-like family, galectins family, integrin superfamily, as well as pigment epithelium derived factor, hepatocyte growth factor, angiopoietins, endothelins, hypoxia-inducible factors, insulin-like growth factors, cytokines, -

Growth Hormone Agents Medical Policy No
Growth Hormone Agents Medical policy no. 30.10.00-1 Effective Date: July 1, 2018 Background: Human growth hormone, also known as somatropin, is produced in the anterior lobe of the pituitary gland. This hormone plays an important role in growth, metabolism, and maintenance of body fat, muscle and bone. Medical necessity Drug Medical Necessity Genotropin® Somatotropin may be considered medically necessary when used for: Humatrope® Norditropin® Children/adolescents with the following: Nutropin®/Nutropin AQ® Neonatal Hypoglycemia Omnitrope® Growth Hormone Deficiency Saizen® Genetic disease with Primary Effects on Growth Serostim® Small for Gestational Age Zomacton® Growth Failure associated with Chronic Renal Insufficiency Zorbtive® Adults with the following: Growth Hormone Deficiency Prader-Willi Syndrome Human Immunodeficiency Virus (HIV)-Related Wasting or Cachexia Short Bowel Syndrome *Preferred growth hormone agents: Genotropin and Norditropin Clinical policy: Clinical Criteria Neonatal Hypoglycemia 1. Diagnosis of ONE of the following: a. Less than (<) 4 months of age with growth deficiency b. History of neonatal hypoglycemia associated with pituitary disease c. Panhypopituitarism 2. Prescribed by or in consultation with an endocrinologist or neonatalogist Growth Hormone Deficiency (Peds) 1. All of the following: a. Diagnosis of pediatric GH deficiency as confirmed by one of the following: i. Projected height is > 2.0 standard deviations [SD] below mid-parental height Policy: Growth Hormones Medical Policy No. 30.10.00-1 Last Updated 08/16/2017 1 ii. Height is > 2.25 SD below population mean iii. Growth velocity is > 2 SD below mean iv. Delayed skeletal maturation of > 2 SD below mean b. One of the following: i. Both of the following: 1. -

Insulin-Like Growth Factor-I
European Journal of Endocrinology (2003) 148 S15–S20 ISSN 0804-4643 Insulin-like growth factor-I: marker for diagnosis of acromegaly and monitoring the efficacy of treatment Georg Brabant Department of Clinical Endocrinology, Hannover Medical School, Hannover, Germany (Correspondence should be addressed to G Brabant, Department of Clinical Endocrinology of the Medizinische Hochschule Hannover, Carl-Neuberg Str.1, 30168 Hannover, Germany; Email: [email protected]) Abstract Acromegaly is caused by chronic excess secretion of growth hormone (GH) and resultant persistent elevation in concentrations of insulin-like growth factor-I (IGF-I), also called somatomedin-C. A number of diagnostic tests are available to support the diagnosis of acromegaly, but those that rely on measurement of serum GH concentrations have important limitations. Concentrations of serum IGF-I, which is produced principally in the liver and mediates the actions of GH, have been shown to correlate with clinical and metabolic markers of disease activity. Addition- ally, normalisation of IGF-I levels in acromegaly is associated with the resolution of symptoms and normal life expectancy. Thus, serum IGF-I is an important marker of disease activity and a sensitive, practical, and reliable measure of integrated GH concentrations in patients with acromegaly. European Journal of Endocrinology 148 S15–S20 Introduction are bound to IGF-specific binding proteins (IGFBPs), six of which have been characterised (8). The most Acromegaly is caused by excessive secretion of growth abundant of these is IGFBP-3, which forms a ternary hormone (GH; also called somatotropin) and a resul- complex with IGF-I and another protein, the acid- tant persistent elevation of insulin-like growth factor-I labile subunit (ALS). -

Probe Set Name Symbol 1598 G at Growth Arres
Supplementary Table 2. List of stroma related genes (i.e. probe sets overexpressed in core relative to FNA biopsies of the same cancer) Probe set Name Symbol 1598_g_at growth arrest-specific 6 GAS6 200048_s_at jumping translocation breakpoint JTB 200054_at zinc finger protein 259 ZNF259 200055_at TAF10 RNA polymerase II, TATA box binding protein (TBP)-associatedTAF10 factor, 30kDa 200059_s_at ras homolog gene family, member A RHOA 200060_s_at RNA binding protein S1, serine-rich domain RNPS1 200070_at chromosome 2 open reading frame 24 C2orf24 200613_at adaptor-related protein complex 2, mu 1 subunit AP2M1 200663_at CD63 molecule CD63 200665_s_at secreted protein, acidic, cysteine-rich (osteonectin) SPARC 200671_s_at spectrin, beta, non-erythrocytic 1 SPTBN1 200696_s_at gelsolin (amyloidosis, Finnish type) GSN 200704_at lipopolysaccharide-induced TNF factor LITAF 200738_s_at phosphoglycerate kinase 1 PGK1 200760_s_at ADP-ribosylation-like factor 6 interacting protein 5 ARL6IP5 200762_at dihydropyrimidinase-like 2 DPYSL2 200770_s_at laminin, gamma 1 (formerly LAMB2) LAMC1 200771_at laminin, gamma 1 (formerly LAMB2) LAMC1 200772_x_at prothymosin, alpha PTMA 200778_s_at septin 2 2-Sep 200782_at annexin A5 ANXA5 200784_s_at low density lipoprotein-related protein 1 (alpha-2-macroglobulin receptor)LRP1 200785_s_at low density lipoprotein-related protein 1 (alpha-2-macroglobulin receptor)LRP1 200795_at SPARC-like 1 (hevin) SPARCL1 200799_at heat shock 70kDa protein 1A HSPA1A 200807_s_at heat shock 60kDa protein 1 (chaperonin) HSPD1 200811_at