Bacterial Communities Associated with the Hindgut of Tipula Abdominalis Larvae
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mining Diversity of the Natural Biorefinery Housed Within Tipula Abdominalis Larvae for Use in an Industrial Biorefinery For
Insect Science (2010) 13, 303–312, DOI 10.1111/j.1744-7917.2010.01343.x ORIGINAL ARTICLE Mining diversity of the natural biorefinery housed within Tipula abdominalis larvae for use in an industrial biorefinery for production of lignocellulosic ethanol Dana M. Cook and Joy Doran-Peterson Microbiology Department, University of Georgia, Athens, Georgia, USA Abstract Although they are the largest taxonomic group of animals, relatively few in- sects have been examined for symbiotic relationships with micro-organisms. However, this is rapidly changing because of the potential for examination of the natural insect– microbe–lignocellulose interactions to provide insights for biofuel technology. Micro- organisms associated with lignocellulose-consuming insects often facilitate the digestion of the recalcitrant plant diet; therefore these microbial communities may be mined for novel lignocellulose-degrading microbes, or for robust and inexpensive biocatalysts necessary for economically feasible biofuel production from lignocellulose. These insect–microbe interactions are influenced by the ecosystem and specific lignocellulose diet, and appre- ciating the whole ecosystem–insect–microbiota–lignocellulose as a natural biorefinery provides a rich and diverse framework from which to design novel industrial processes. One such natural biorefinery, the Tipula abdominalis larvae in riparian ecosystems, is reviewed herein with applications for biochemical processes and overcoming challenges involved in conversion of lignocellulosic biomass to fuel ethanol. From the dense and diverse T. abdominalis larval hindgut microbial community, a cellulolytic bacterial iso- late, 27C64, demonstrated enzymatic activity toward many model plant polymers and also produced a bacterial antibiotic. 27C64 was co-cultured with yeast in fermentation of pine to ethanol, which allowed for a 20% reduction of commercial enzyme. -

Table S1. Bacterial Otus from 16S Rrna
Table S1. Bacterial OTUs from 16S rRNA sequencing analysis including only taxa which were identified to genus level (those OTUs identified as Ambiguous taxa, uncultured bacteria or without genus-level identifications were omitted). OTUs with only a single representative across all samples were also omitted. Taxa are listed from most to least abundant. Pitcher Plant Sample Class Order Family Genus CB1p1 CB1p2 CB1p3 CB1p4 CB5p234 Sp3p2 Sp3p4 Sp3p5 Sp5p23 Sp9p234 sum Gammaproteobacteria Legionellales Coxiellaceae Rickettsiella 1 2 0 1 2 3 60194 497 1038 2 61740 Alphaproteobacteria Rhodospirillales Rhodospirillaceae Azospirillum 686 527 10513 485 11 3 2 7 16494 8201 36929 Sphingobacteriia Sphingobacteriales Sphingobacteriaceae Pedobacter 455 302 873 103 16 19242 279 55 760 1077 23162 Betaproteobacteria Burkholderiales Oxalobacteraceae Duganella 9060 5734 2660 40 1357 280 117 29 129 35 19441 Gammaproteobacteria Pseudomonadales Pseudomonadaceae Pseudomonas 3336 1991 3475 1309 2819 233 1335 1666 3046 218 19428 Betaproteobacteria Burkholderiales Burkholderiaceae Paraburkholderia 0 1 0 1 16051 98 41 140 23 17 16372 Sphingobacteriia Sphingobacteriales Sphingobacteriaceae Mucilaginibacter 77 39 3123 20 2006 324 982 5764 408 21 12764 Gammaproteobacteria Pseudomonadales Moraxellaceae Alkanindiges 9 10 14 7 9632 6 79 518 1183 65 11523 Betaproteobacteria Neisseriales Neisseriaceae Aquitalea 0 0 0 0 1 1577 5715 1471 2141 177 11082 Flavobacteriia Flavobacteriales Flavobacteriaceae Flavobacterium 324 219 8432 533 24 123 7 15 111 324 10112 Alphaproteobacteria -

Durham E-Theses
Durham E-Theses The feeding ecology of certain larvae in the genus tipula (Tipulidae, Diptera), with special reference to their utilisation of Bryophytes Todd, Catherine Mary How to cite: Todd, Catherine Mary (1993) The feeding ecology of certain larvae in the genus tipula (Tipulidae, Diptera), with special reference to their utilisation of Bryophytes, Durham theses, Durham University. Available at Durham E-Theses Online: http://etheses.dur.ac.uk/5699/ Use policy The full-text may be used and/or reproduced, and given to third parties in any format or medium, without prior permission or charge, for personal research or study, educational, or not-for-prot purposes provided that: • a full bibliographic reference is made to the original source • a link is made to the metadata record in Durham E-Theses • the full-text is not changed in any way The full-text must not be sold in any format or medium without the formal permission of the copyright holders. Please consult the full Durham E-Theses policy for further details. Academic Support Oce, Durham University, University Oce, Old Elvet, Durham DH1 3HP e-mail: [email protected] Tel: +44 0191 334 6107 http://etheses.dur.ac.uk 2 THE FEEDING ECOLOGY OF CERTAIN LARVAE IN THE GENUS TIPULA (TIPULIDAE, DIPTERA), WITH SPECIAL REFERENCE TO THEIR UTILISATION OF BRYOPHYTES Catherine Mary Todd B.Sc. (London), M.Sc. (Durham) The copyright of this thesis rests with the author. No quotation from it should be published without his prior written consent and information derived from it should be acknowledged. A thesis presented in candidature for the degree of Doctor of Philosophy in the University of Durham, 1993 FEB t99^ Abstract Bryophytes are rarely used as a food source by any animal species, but the genus Tipula (Diptera, Tipulidae) contains some of the few insect species able to feed, and complete their life-cycle, on bryophytes. -

Analysis of Cellulolytic and Hemicellulolytic Enzyme Activity
Insect Science (2010) 17, 291–302, DOI 10.1111/j.1744-7917.2010.01346.x ORIGINAL ARTICLE Analysis of cellulolytic and hemicellulolytic enzyme activity within the Tipula abdominalis (Diptera: Tipulidae) larval gut and characterization of Crocebacterium ilecola gen. nov., sp. nov., isolated from the Tipula abdominalis larval hindgut Theresa E. Rogers and Joy Doran-Peterson Microbiology Department, University of Georgia, Athens, Georgia, USA Abstract In forested stream ecosystems of the north and eastern United States, larvae of the aquatic crane fly Tipula abdominalis are important shredders of leaf litter detritus. T. abdominalis larvae harbor a dense and diverse microbial community in their hindgut that may aide in the degradation of lignocellulose. In this study, the activities of cellulolytic and hemicellulolytic enzymes were demonstrated from hindgut extracts and from bacterial isolates using model sugar substrates. One of the bacterial isolates was further characterized as a member of the family Microbacteriaceae. Taxonomic position of the isolate within this family was determined by a polyphasic approach, as is commonly employed for the separation of genera within the family Microbacteriaceae. The bacterial isolate is Gram- type positive, motile, non-sporulating, and rod-shaped. The G + C content of the DNA is 64.9 mol%. The cell wall contains B2γ type peptidoglycan, D- and L-diaminobutyric acid as the diamino acid, and rhamnose as the predominant sugar. The predominant fatty acids are 12-methyltetradecanoic acid (ai-C15:0) and 14-methylhexadecanoic acid (ai-C17:0). The isolate forms a distinct lineage within the family Microbacteriaceae, as determined by 16S rRNA sequence analysis. We propose the name Crocebacterium ilecola gen. -

Diverse Allochthonous Resource Quality Effects on Headwater Stream Communities Through Insect-Microbe Interactions
DIVERSE ALLOCHTHONOUS RESOURCE QUALITY EFFECTS ON HEADWATER STREAM COMMUNITIES THROUGH INSECT-MICROBE INTERACTIONS By Courtney Larson A DISSERTATION Submitted to Michigan State University in partial fulfillment of the requirements for the degree of Entomology—Doctor of Philosophy Ecology, Evolutionary Biology and Behavior—Dual Major 2020 ABSTRACT DIVERSE ALLOCHTHONOUS RESOURCE QUALITY EFFECTS ON HEADWATER STREAM COMMUNITIES THROUGH INSECT-MICROBE INTERACTIONS By Courtney Larson Freshwater resources are vital to environmental sustainability and human health; yet, they are inundated by multiple stressors, leaving aquatic communities to face unknown consequences. Headwater streams are highly reliant on allochthonous sources of energy. Riparian trees shade the stream, limiting primary production, causing macroinvertebrates to consume an alternative food source. Traditionally, leaf litter fallen from riparian trees is the primary allochthonous resource, but other sources, such as salmon carrion associated with annual salmon runs, may also be important. An alteration in the quantity or quality of these sources may have far reaching effects not only on the organisms that directly consume the allochthonous resource (shredders), but also on other functional feeding groups. Allochthonous resources directly and indirectly change stream microbial communities, which are used by consumers with potential changes to their life histories and behavior traits. The objective of my research was to determine the influence allochthonous resources have on stream -

Biodiversité Du Microbiome Cutané Des Organismes Marins : Variabilité, Déterminants Et Importance Dans L’Écosystème Marlène Chiarello
Biodiversité du microbiome cutané des organismes marins : variabilité, déterminants et importance dans l’écosystème Marlène Chiarello To cite this version: Marlène Chiarello. Biodiversité du microbiome cutané des organismes marins : variabilité, détermi- nants et importance dans l’écosystème. Microbiologie et Parasitologie. Université Montpellier, 2017. Français. NNT : 2017MONTT092. tel-01693132 HAL Id: tel-01693132 https://tel.archives-ouvertes.fr/tel-01693132 Submitted on 25 Jan 2018 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. ! ! ! Délivré par l’Université de Montpellier Préparée au sein de l’école doctorale GAIA Et de l’unité de recherche MARBEC (Centre pour la Biodiversité marine, l’Exploitation et la Conservation) Spécialité : Écologie Fonctionnelle et Sciences Agronomiques Présentée par Marlène Chiarello Biodiversité du microbiome cutané des organismes marins : variabilité, déterminants et importance dans l’écosystème Soutenue le 29 novembre 2017 devant le jury composé de M. Thierry BOUVIER, DR, CNRS Directeur M. Frédéric DELSUC, DR, CNRS Examinateur M. Pierre GALAND, DR, CNRS Invité M. Fabrice NOT, DR, CNRS Invité M. Loïc PELLISIER, Asst. Prof., ETH Zürich Rapporteur M. Télesphore SIME-NGANDO, DR, CNRS Rapporteur Mme Eve TOULZA, MCF, Université de Perpignan Examinateur M. -
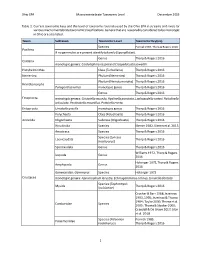
Ohio EPA Macroinvertebrate Taxonomic Level December 2019 1 Table 1. Current Taxonomic Keys and the Level of Taxonomy Routinely U
Ohio EPA Macroinvertebrate Taxonomic Level December 2019 Table 1. Current taxonomic keys and the level of taxonomy routinely used by the Ohio EPA in streams and rivers for various macroinvertebrate taxonomic classifications. Genera that are reasonably considered to be monotypic in Ohio are also listed. Taxon Subtaxon Taxonomic Level Taxonomic Key(ies) Species Pennak 1989, Thorp & Rogers 2016 Porifera If no gemmules are present identify to family (Spongillidae). Genus Thorp & Rogers 2016 Cnidaria monotypic genera: Cordylophora caspia and Craspedacusta sowerbii Platyhelminthes Class (Turbellaria) Thorp & Rogers 2016 Nemertea Phylum (Nemertea) Thorp & Rogers 2016 Phylum (Nematomorpha) Thorp & Rogers 2016 Nematomorpha Paragordius varius monotypic genus Thorp & Rogers 2016 Genus Thorp & Rogers 2016 Ectoprocta monotypic genera: Cristatella mucedo, Hyalinella punctata, Lophopodella carteri, Paludicella articulata, Pectinatella magnifica, Pottsiella erecta Entoprocta Urnatella gracilis monotypic genus Thorp & Rogers 2016 Polychaeta Class (Polychaeta) Thorp & Rogers 2016 Annelida Oligochaeta Subclass (Oligochaeta) Thorp & Rogers 2016 Hirudinida Species Klemm 1982, Klemm et al. 2015 Anostraca Species Thorp & Rogers 2016 Species (Lynceus Laevicaudata Thorp & Rogers 2016 brachyurus) Spinicaudata Genus Thorp & Rogers 2016 Williams 1972, Thorp & Rogers Isopoda Genus 2016 Holsinger 1972, Thorp & Rogers Amphipoda Genus 2016 Gammaridae: Gammarus Species Holsinger 1972 Crustacea monotypic genera: Apocorophium lacustre, Echinogammarus ischnus, Synurella dentata Species (Taphromysis Mysida Thorp & Rogers 2016 louisianae) Crocker & Barr 1968; Jezerinac 1993, 1995; Jezerinac & Thoma 1984; Taylor 2000; Thoma et al. Cambaridae Species 2005; Thoma & Stocker 2009; Crandall & De Grave 2017; Glon et al. 2018 Species (Palaemon Pennak 1989, Palaemonidae kadiakensis) Thorp & Rogers 2016 1 Ohio EPA Macroinvertebrate Taxonomic Level December 2019 Taxon Subtaxon Taxonomic Level Taxonomic Key(ies) Informal grouping of the Arachnida Hydrachnidia Smith 2001 water mites Genus Morse et al. -

Evolutionary Background Entities at the Cellular and Subcellular Levels in Bodies of Invertebrate Animals
The Journal of Theoretical Fimpology Volume 2, Issue 4: e-20081017-2-4-14 December 28, 2014 www.fimpology.com Evolutionary Background Entities at the Cellular and Subcellular Levels in Bodies of Invertebrate Animals Shu-dong Yin Cory H. E. R. & C. Inc. Burnaby, British Columbia, Canada Email: [email protected] ________________________________________________________________________ Abstract The novel recognition that individual bodies of normal animals are actually inhabited by subcellular viral entities and membrane-enclosed microentities, prokaryotic bacterial and archaeal cells and unicellular eukaryotes such as fungi and protists has been supported by increasing evidences since the emergence of culture-independent approaches. However, how to understand the relationship between animal hosts including human beings and those non-host microentities or microorganisms is challenging our traditional understanding of pathogenic relationship in human medicine and veterinary medicine. In recent novel evolution theories, the relationship between animals and their environments has been deciphered to be the interaction between animals and their environmental evolutionary entities at the same and/or different evolutionary levels;[1-3] and evolutionary entities of the lower evolutionary levels are hypothesized to be the evolutionary background entities of entities at the higher evolutionary levels.[1,2] Therefore, to understand the normal existence of microentities or microorganisms in multicellular animal bodies is becoming the first priority for elucidating the ecological and evolutiological relationships between microorganisms and nonhuman macroorganisms. The evolutionary background entities at the cellular and subcellular levels in bodies of nonhuman vertebrate animals have been summarized recently.[4] In this paper, the author tries to briefly review the evolutionary background entities (EBE) at the cellular and subcellular levels for several selected invertebrate animal species. -

The Role of the Surface Smear Microbiome in the Development of Defective Smear on Surface-Ripened Red-Smear Cheese
AIMS Microbiology, 4(4): 622–641. DOI: 10.3934/microbiol.2018.4.622 Received: 29 June 2018 Accepted: 20 September 2018 Published: 25 October 2018 http://www.aimspress.com/journal/microbiology Research article The role of the surface smear microbiome in the development of defective smear on surface-ripened red-smear cheese Jasmine S. Ritschard1,†, Lea Amato2,†, Yadhu Kumar3, Britta Müller3, Leo Meile2 and Markus Schuppler1,* 1 Laboratory of Food Microbiology, Institute of Food, Nutrition and Health, ETH Zurich, Schmelzbergstrasse 7, 8092 Zurich, Switzerland 2 Laboratory of Food Biotechnology, Institute of Food, Nutrition and Health, ETH Zurich, Schmelzbergstrasse 7, 8092 Zurich, Switzerland 3 Eurofins GATC Biotech AG, Jakob-Stadler-Platz 7, 78467 Konstanz, Germany * Correspondence: Email: [email protected]; Tel: +41446327862; Fax: +41446321266. † These two authors contributed equally. Abstract: The complex smear microbiota colonizing the surface of red-smear cheese fundamentally impacts the ripening process, appearance and shelf life of cheese. To decipher the prokaryotic composition of the cheese smear microbiome, the surface of a semi-hard surface ripened cheese was studied post-ripening by culture-based and culture-independent molecular approaches. The aim was to detect potential bacterial alterations in the composition of the cheese smear microbiota resulting from cheese storage in vacuum film-prepackaging, which is often accompanied by the development of a surface smear defect. Next-generation sequencing of amplified 16S rRNA gene fragments revealed an unexpected high diversity of a total of 132 different genera from the domains Bacteria and Archaea on the cheese surface. Beside typical smear organisms, our study revealed the presence of several microorganisms so far not associated with cheese, but related to milk, farm and cheese dairy environments. -

Journal of Cave and Karst Studies
June 2020 Volume 82, Number 2 JOURNAL OF ISSN 1090-6924 A Publication of the National CAVE AND KARST Speleological Society STUDIES DEDICATED TO THE ADVANCEMENT OF SCIENCE, EDUCATION, EXPLORATION, AND CONSERVATION Published By BOARD OF EDITORS The National Speleological Society Anthropology George Crothers http://caves.org/pub/journal University of Kentucky Lexington, KY Office [email protected] 6001 Pulaski Pike NW Huntsville, AL 35810 USA Conservation-Life Sciences Julian J. Lewis & Salisa L. Lewis Tel:256-852-1300 Lewis & Associates, LLC. [email protected] Borden, IN [email protected] Editor-in-Chief Earth Sciences Benjamin Schwartz Malcolm S. Field Texas State University National Center of Environmental San Marcos, TX Assessment (8623P) [email protected] Office of Research and Development U.S. Environmental Protection Agency Leslie A. North 1200 Pennsylvania Avenue NW Western Kentucky University Bowling Green, KY Washington, DC 20460-0001 [email protected] 703-347-8601 Voice 703-347-8692 Fax [email protected] Mario Parise University Aldo Moro Production Editor Bari, Italy [email protected] Scott A. Engel Knoxville, TN Carol Wicks 225-281-3914 Louisiana State University [email protected] Baton Rouge, LA [email protected] Exploration Paul Burger National Park Service Eagle River, Alaska [email protected] Microbiology Kathleen H. Lavoie State University of New York Plattsburgh, NY [email protected] Paleontology Greg McDonald National Park Service Fort Collins, CO The Journal of Cave and Karst Studies , ISSN 1090-6924, CPM [email protected] Number #40065056, is a multi-disciplinary, refereed journal pub- lished four times a year by the National Speleological Society. -

Proposed Minimal Standards for Describing New Genera and Species of the Suborder Micrococcineae
International Journal of Systematic and Evolutionary Microbiology (2009), 59, 1823–1849 DOI 10.1099/ijs.0.012971-0 Proposed minimal standards for describing new genera and species of the suborder Micrococcineae Peter Schumann,1 Peter Ka¨mpfer,2 Hans-Ju¨rgen Busse 3 and Lyudmila I. Evtushenko4 for the Subcommittee on the Taxonomy of the Suborder Micrococcineae of the International Committee on Systematics of Prokaryotes Correspondence 1DSMZ-Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH, Inhoffenstraße 7B, P. Schumann 38124 Braunschweig, Germany [email protected] 2Institut fu¨r Angewandte Mikrobiologie, Justus-Liebig-Universita¨t, 35392 Giessen, Germany 3Institut fu¨r Bakteriologie, Mykologie und Hygiene, Veterina¨rmedizinische Universita¨t, A-1210 Wien, Austria 4All-Russian Collection of Microorganisms (VKM), G. K. Skryabin Institute of Biochemistry and Physiology of Microorganisms, RAS, Pushchino, Moscow Region 142290, Russia The Subcommittee on the Taxonomy of the Suborder Micrococcineae of the International Committee on Systematics of Prokaryotes has agreed on minimal standards for describing new genera and species of the suborder Micrococcineae. The minimal standards are intended to provide bacteriologists involved in the taxonomy of members of the suborder Micrococcineae with a set of essential requirements for the description of new taxa. In addition to sequence data comparisons of 16S rRNA genes or other appropriate conservative genes, phenotypic and genotypic criteria are compiled which are considered essential for the comprehensive characterization of new members of the suborder Micrococcineae. Additional features are recommended for the characterization and differentiation of genera and species with validly published names. INTRODUCTION Aureobacterium and Rothia/Stomatococcus) and one pair of homotypic synonyms (Pseudoclavibacter/Zimmer- The suborder Micrococcineae was established by mannella) (Table 1 and http://www.the-icsp.org/taxa/ Stackebrandt et al. -

Aquatic Insects: Holometabola – Diptera, Suborder Nematocera
Glime, J. M. 2017. Aquatic Insects: Holometabola – Diptera, Suborder Nematocera. Chapt. 11-13a. In: Glime, J. M. 11-13a-1 Bryophyte Ecology. Volume 2. Bryological Interaction. Ebook sponsored by Michigan Technological University and the International Association of Bryologists. Last updated 19 July 2020 and available at <http://digitalcommons.mtu.edu/bryophyte-ecology2/>. CHAPTER 11-13a AQUATIC INSECTS: HOLOMETABOLA – DIPTERA, SUBORDER NEMATOCERA TABLE OF CONTENTS DIPTERA – Flies .......................................................................................................................................... 11-13a-2 Suborder Nematocera ............................................................................................................................. 11-13a-5 Nymphomyiidae .............................................................................................................................. 11-13a-6 Cylindrotomidae – Long-bodied Craneflies .................................................................................... 11-13a-6 Limoniidae – Limoniid Craneflies .................................................................................................. 11-13a-8 Pediciidae – Hairy-eyed Craneflies ............................................................................................... 11-13a-11 Tipulidae – Craneflies ................................................................................................................... 11-13a-11 Anisopodidae – Wood Gnats, Window Gnats .............................................................................