Systematic Identification of Factors for Provirus Silencing in Embryonic Stem Cells
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Regulation of Oxidized Base Damage Repair by Chromatin Assembly Factor 1 Subunit a Chunying Yang1,*,†, Shiladitya Sengupta1,2,*,†, Pavana M
Published online 27 October 2016 Nucleic Acids Research, 2017, Vol. 45, No. 2 739–748 doi: 10.1093/nar/gkw1024 Regulation of oxidized base damage repair by chromatin assembly factor 1 subunit A Chunying Yang1,*,†, Shiladitya Sengupta1,2,*,†, Pavana M. Hegde1,JoyMitra1, Shuai Jiang3, Brooke Holey3, Altaf H. Sarker3, Miaw-Sheue Tsai3, Muralidhar L. Hegde1,2,4 and Sankar Mitra1,2,* 1Department of Radiation Oncology, Houston Methodist Research Institute, Houston, TX 77030, USA, 2Weill Cornell Medical College, Cornell University, New York, NY 10065, USA, 3Biological Systems and Engineering Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA and 4Houston Methodist Neurological Institute, Houston, TX 77030, USA Received March 23, 2016; Revised October 13, 2016; Editorial Decision October 17, 2016; Accepted October 19, 2016 ABSTRACT INTRODUCTION Reactive oxygen species (ROS), generated both en- ROS, continuously generated in mammalian cells both en- dogenously and in response to exogenous stress, in- dogenously and by environmental genotoxicants, induce duce point mutations by mis-replication of oxidized various genomic lesions, including oxidized bases, abasic bases and other lesions in the genome. Repair of (AP) sites and single-strand breaks (SSBs). If unrepaired or these lesions via base excision repair (BER) pathway mis-repaired, DNA lesions would cause mutations which may lead to cytotoxicity and cell death and also carcino- maintains genomic fidelity. Regulation of the BER genic transformation (1). The base excision repair (BER) pathway for mutagenic oxidized bases, initiated by pathway, responsible for repair of oxidized base lesions NEIL1 and other DNA glycosylases at the chromatin which contribute to drug/radiation sensitivity is highly con- level remains unexplored. -

Screening and Identification of Hub Genes in Bladder Cancer by Bioinformatics Analysis and KIF11 Is a Potential Prognostic Biomarker
ONCOLOGY LETTERS 21: 205, 2021 Screening and identification of hub genes in bladder cancer by bioinformatics analysis and KIF11 is a potential prognostic biomarker XIAO‑CONG MO1,2*, ZI‑TONG ZHANG1,3*, MENG‑JIA SONG1,2, ZI‑QI ZHOU1,2, JIAN‑XIONG ZENG1,2, YU‑FEI DU1,2, FENG‑ZE SUN1,2, JIE‑YING YANG1,2, JUN‑YI HE1,2, YUE HUANG1,2, JIAN‑CHUAN XIA1,2 and DE‑SHENG WENG1,2 1State Key Laboratory of Oncology in South China, Collaborative Innovation Centre for Cancer Medicine; 2Department of Biotherapy, Sun Yat‑Sen University Cancer Center; 3Department of Radiation Oncology, Sun Yat‑Sen University Cancer Center, Guangzhou, Guangdong 510060, P.R. China Received July 31, 2020; Accepted December 18, 2020 DOI: 10.3892/ol.2021.12466 Abstract. Bladder cancer (BC) is the ninth most common immunohistochemistry and western blotting. In summary, lethal malignancy worldwide. Great efforts have been devoted KIF11 was significantly upregulated in BC and might act as to clarify the pathogenesis of BC, but the underlying molecular a potential prognostic biomarker. The present identification mechanisms remain unclear. To screen for the genes associated of DEGs and hub genes in BC may provide novel insight for with the progression and carcinogenesis of BC, three datasets investigating the molecular mechanisms of BC. were obtained from the Gene Expression Omnibus. A total of 37 tumor and 16 non‑cancerous samples were analyzed to Introduction identify differentially expressed genes (DEGs). Subsequently, 141 genes were identified, including 55 upregulated and Bladder cancer (BC) is the ninth most common malignancy 86 downregulated genes. The protein‑protein interaction worldwide with substantial morbidity and mortality. -

Genetic Drivers of Pancreatic Islet Function
| INVESTIGATION Genetic Drivers of Pancreatic Islet Function Mark P. Keller,*,1 Daniel M. Gatti,†,1 Kathryn L. Schueler,* Mary E. Rabaglia,* Donnie S. Stapleton,* Petr Simecek,† Matthew Vincent,† Sadie Allen,‡ Aimee Teo Broman,§ Rhonda Bacher,§ Christina Kendziorski,§ Karl W. Broman,§ Brian S. Yandell,** Gary A. Churchill,†,2 and Alan D. Attie*,2 *Department of Biochemistry, §Department of Biostatistics and Medical Informatics, and **Department of Horticulture, University of Wisconsin–Madison, Wisconsin 53706-1544, †The Jackson Laboratory, Bar Harbor, Maine 06409, and ‡Maine School of Science and Mathematics, Limestone, Maine 06409, ORCID IDs: 0000-0002-7405-5552 (M.P.K.); 0000-0002-4914-6671 (K.W.B.); 0000-0001-9190-9284 (G.A.C.); 0000-0002-0568-2261 (A.D.A.) ABSTRACT The majority of gene loci that have been associated with type 2 diabetes play a role in pancreatic islet function. To evaluate the role of islet gene expression in the etiology of diabetes, we sensitized a genetically diverse mouse population with a Western diet high in fat (45% kcal) and sucrose (34%) and carried out genome-wide association mapping of diabetes-related phenotypes. We quantified mRNA abundance in the islets and identified 18,820 expression QTL. We applied mediation analysis to identify candidate causal driver genes at loci that affect the abundance of numerous transcripts. These include two genes previously associated with monogenic diabetes (PDX1 and HNF4A), as well as three genes with nominal association with diabetes-related traits in humans (FAM83E, IL6ST, and SAT2). We grouped transcripts into gene modules and mapped regulatory loci for modules enriched with transcripts specific for a-cells, and another specific for d-cells. -

Supplementary Data
SUPPLEMENTARY DATA A cyclin D1-dependent transcriptional program predicts clinical outcome in mantle cell lymphoma Santiago Demajo et al. 1 SUPPLEMENTARY DATA INDEX Supplementary Methods p. 3 Supplementary References p. 8 Supplementary Tables (S1 to S5) p. 9 Supplementary Figures (S1 to S15) p. 17 2 SUPPLEMENTARY METHODS Western blot, immunoprecipitation, and qRT-PCR Western blot (WB) analysis was performed as previously described (1), using cyclin D1 (Santa Cruz Biotechnology, sc-753, RRID:AB_2070433) and tubulin (Sigma-Aldrich, T5168, RRID:AB_477579) antibodies. Co-immunoprecipitation assays were performed as described before (2), using cyclin D1 antibody (Santa Cruz Biotechnology, sc-8396, RRID:AB_627344) or control IgG (Santa Cruz Biotechnology, sc-2025, RRID:AB_737182) followed by protein G- magnetic beads (Invitrogen) incubation and elution with Glycine 100mM pH=2.5. Co-IP experiments were performed within five weeks after cell thawing. Cyclin D1 (Santa Cruz Biotechnology, sc-753), E2F4 (Bethyl, A302-134A, RRID:AB_1720353), FOXM1 (Santa Cruz Biotechnology, sc-502, RRID:AB_631523), and CBP (Santa Cruz Biotechnology, sc-7300, RRID:AB_626817) antibodies were used for WB detection. In figure 1A and supplementary figure S2A, the same blot was probed with cyclin D1 and tubulin antibodies by cutting the membrane. In figure 2H, cyclin D1 and CBP blots correspond to the same membrane while E2F4 and FOXM1 blots correspond to an independent membrane. Image acquisition was performed with ImageQuant LAS 4000 mini (GE Healthcare). Image processing and quantification were performed with Multi Gauge software (Fujifilm). For qRT-PCR analysis, cDNA was generated from 1 µg RNA with qScript cDNA Synthesis kit (Quantabio). qRT–PCR reaction was performed using SYBR green (Roche). -

Variation in Protein Coding Genes Identifies Information
bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 1 1 2 3 4 5 Variation in protein coding genes identifies information flow as a contributor to 6 animal complexity 7 8 Jack Dean, Daniela Lopes Cardoso and Colin Sharpe* 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 Institute of Biological and Biomedical Sciences 25 School of Biological Science 26 University of Portsmouth, 27 Portsmouth, UK 28 PO16 7YH 29 30 * Author for correspondence 31 [email protected] 32 33 Orcid numbers: 34 DLC: 0000-0003-2683-1745 35 CS: 0000-0002-5022-0840 36 37 38 39 40 41 42 43 44 45 46 47 48 49 Abstract bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 2 1 Across the metazoans there is a trend towards greater organismal complexity. How 2 complexity is generated, however, is uncertain. Since C.elegans and humans have 3 approximately the same number of genes, the explanation will depend on how genes are 4 used, rather than their absolute number. -

Differential Requirements for Tousled-Like Kinases 1 and 2 in Mammalian Development
Cell Death and Differentiation (2017) 24, 1872–1885 & 2017 Macmillan Publishers Limited, part of Springer Nature. All rights reserved 1350-9047/17 www.nature.com/cdd Differential requirements for Tousled-like kinases 1 and 2 in mammalian development Sandra Segura-Bayona1,8, Philip A Knobel1,8, Helena González-Burón1,8, Sameh A Youssef2,3, Aida Peña-Blanco1, Étienne Coyaud4,5, Teresa López-Rovira1, Katrin Rein1, Lluís Palenzuela1, Julien Colombelli1, Stephen Forrow1, Brian Raught4,5, Anja Groth6, Alain de Bruin2,7 and Travis H Stracker*,1 The regulation of chromatin structure is critical for a wide range of essential cellular processes. The Tousled-like kinases, TLK1 and TLK2, regulate ASF1, a histone H3/H4 chaperone, and likely other substrates, and their activity has been implicated in transcription, DNA replication, DNA repair, RNA interference, cell cycle progression, viral latency, chromosome segregation and mitosis. However, little is known about the functions of TLK activity in vivo or the relative functions of the highly similar TLK1 and TLK2 in any cell type. To begin to address this, we have generated Tlk1- and Tlk2-deficient mice. We found that while TLK1 was dispensable for murine viability, TLK2 loss led to late embryonic lethality because of placental failure. TLK2 was required for normal trophoblast differentiation and the phosphorylation of ASF1 was reduced in placentas lacking TLK2. Conditional bypass of the placental phenotype allowed the generation of apparently healthy Tlk2-deficient mice, while only the depletion of both TLK1 and TLK2 led to extensive genomic instability, indicating that both activities contribute to genome maintenance. Our data identifies a specific role for TLK2 in placental function during mammalian development and suggests that TLK1 and TLK2 have largely redundant roles in genome maintenance. -

Histone Chaperone CHAF1A Inhibits Differentiation and Promotes Aggressive Neuroblastoma
Published OnlineFirst December 12, 2013; DOI: 10.1158/0008-5472.CAN-13-1315 Cancer Molecular and Cellular Pathobiology Research Histone Chaperone CHAF1A Inhibits Differentiation and Promotes Aggressive Neuroblastoma Eveline Barbieri1, Katleen De Preter3, Mario Capasso4, Zaowen Chen1, Danielle M. Hsu2, Gian Paolo Tonini5, Steve Lefever3, John Hicks1, Rogier Versteeg7, Andrea Pession6, Frank Speleman3, Eugene S. Kim2, and Jason M. Shohet1 Abstract Neuroblastoma arises from the embryonal neural crest secondary to a block in differentiation. Long-term patient survival correlates inversely with the extent of differentiation, and treatment with retinoic acid or other prodifferentiation agents improves survival modestly. In this study, we show the histone chaperone and epigenetic regulator CHAF1A functions in maintaining the highly dedifferentiated state of this aggressive malignancy. CHAF1A is a subunit of the chromatin modifier chromatin assembly factor 1 and it regulates H3K9 trimethylation of key target genes regulating proliferation, survival, and differentiation. Elevated CHAF1A expression strongly correlated with poor prognosis. Conversely, CHAF1A loss-of-function was sufficient to drive neuronal differentiation in vitro and in vivo. Transcriptome analysis of cells lacking CHAF1A revealed repression of oncogenic signaling pathways and a normalization of glycolytic metabolism. Our findings demonstrate that CHAF1A restricts neural crest differentiation and contributes to the pathogenesis of high-risk neuroblastoma. Cancer Res; 74(3); 1–10. -

Differential Expression Profile Analysis of DNA Damage Repair Genes in CD133+/CD133‑ Colorectal Cancer Cells
ONCOLOGY LETTERS 14: 2359-2368, 2017 Differential expression profile analysis of DNA damage repair genes in CD133+/CD133‑ colorectal cancer cells YUHONG LU1*, XIN ZHOU2*, QINGLIANG ZENG2, DAISHUN LIU3 and CHANGWU YUE3 1College of Basic Medicine, Zunyi Medical University, Zunyi; 2Deparment of Gastroenterological Surgery, Affiliated Hospital of Zunyi Medical University, Zunyi;3 Zunyi Key Laboratory of Genetic Diagnosis and Targeted Drug Therapy, The First People's Hospital of Zunyi, Zunyi, Guizhou 563003, P.R. China Received July 20, 2015; Accepted January 6, 2017 DOI: 10.3892/ol.2017.6415 Abstract. The present study examined differential expression cells. By contrast, 6 genes were downregulated and none levels of DNA damage repair genes in COLO 205 colorectal were upregulated in the CD133+ cells compared with the cancer cells, with the aim of identifying novel biomarkers for COLO 205 cells. These findings suggest that CD133+ cells the molecular diagnosis and treatment of colorectal cancer. may possess the same DNA repair capacity as COLO 205 COLO 205-derived cell spheres were cultured in serum-free cells. Heterogeneity in the expression profile of DNA damage medium supplemented with cell factors, and CD133+/CD133- repair genes was observed in COLO 205 cells, and COLO cells were subsequently sorted using an indirect CD133 205-derived CD133- cells and CD133+ cells may therefore microbead kit. In vitro differentiation and tumorigenicity assays provide a reference for molecular diagnosis, therapeutic target in BABA/c nude mice were performed to determine whether selection and determination of the treatment and prognosis for the CD133+ cells also possessed stem cell characteristics, in colorectal cancer. -
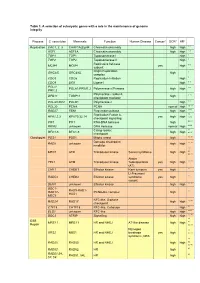
Table 1. a Selection of Eukaryotic Genes with a Role in the Maintenance of Genome Integrity
Table 1. A selection of eukaryotic genes with a role in the maintenance of genome integrity Process S. cerevisiae Mammals Function Human Disease Cancer* GCR* HR* Replication CAC1, 2, 3 CHAF1A,B,p48 Chromatin assembly high high 1, 2 1, 2 ASF1 ASF1A Chromatin assembly high high 3 TOP1 TOP1 Topoisomerase I high 3 TOP2 TOP2 Topoisomerase II high Replicative helicase 4, 5 MCM4 MCM4 yes high subunit Origin Replication 6 ORC3/5 ORC3/6L high complex 7 CDC6 CDC6 Replication Initiation high 7, 8 CDC9 LIG1 Ligase I high POL1/ 7-10 POLA1/PRIM1,2 Polymerase α/Primase high high PRI1,2 Polymerase ε subunit, 6, 11 DPB11 TOBP11 high checkpoint mediator POL3/CDC2 POLD1 Polymerase δ high 7, 8 POL30 PCNA PCNA normal high 12, 13 RAD27 FEN1 Flap endonuclease high high 14-16 Replication Factor A, 14, RFA1,2,3 RPA70,32,14 yes high high 17-19 checkpoint signalling PIF1 PIF1 RNA-DNA helicase high 20, 21 RRM3 unknown DNA Helicase normal high 22-26 Clamp loader, 11, RFC1-5 RFC1-5 high high 20, 27 checkpoint Checkpoint PDS1 PDS1 Mitotic arrest high 11, 28 Damage checkpoint 11, 29 RAD9 unknown high high mediator 11, MEC1 ATR Transducer kinase Seckel syndrome high high 28, 30, 31 Ataxia TEL1 ATM Transducer kinase Telangiectasia yes high high 11, 28 (AT) CHK1 CHEK1 Effector kinase Rare tumours yes high 11 Li-Fraumeni RAD53 CHEK2 Effector kinase syndrome yes high 11 variant DUN1 unknown Effector kinase high high 11, 32 DDC1- RAD9-RAD1- 11 RAD17- PCNA-like complex high HUS1 MEC3 RFC-like, S-phase 11, 33 RAD24 RAD17 high high checkponit 34 CTF18 CHTF18 RFC-like, Cohesion -

The Genetic Program of Pancreatic Beta-Cell Replication in Vivo
Page 1 of 65 Diabetes The genetic program of pancreatic beta-cell replication in vivo Agnes Klochendler1, Inbal Caspi2, Noa Corem1, Maya Moran3, Oriel Friedlich1, Sharona Elgavish4, Yuval Nevo4, Aharon Helman1, Benjamin Glaser5, Amir Eden3, Shalev Itzkovitz2, Yuval Dor1,* 1Department of Developmental Biology and Cancer Research, The Institute for Medical Research Israel-Canada, The Hebrew University-Hadassah Medical School, Jerusalem 91120, Israel 2Department of Molecular Cell Biology, Weizmann Institute of Science, Rehovot, Israel. 3Department of Cell and Developmental Biology, The Silberman Institute of Life Sciences, The Hebrew University of Jerusalem, Jerusalem 91904, Israel 4Info-CORE, Bioinformatics Unit of the I-CORE Computation Center, The Hebrew University and Hadassah, The Institute for Medical Research Israel- Canada, The Hebrew University-Hadassah Medical School, Jerusalem 91120, Israel 5Endocrinology and Metabolism Service, Department of Internal Medicine, Hadassah-Hebrew University Medical Center, Jerusalem 91120, Israel *Correspondence: [email protected] Running title: The genetic program of pancreatic β-cell replication 1 Diabetes Publish Ahead of Print, published online March 18, 2016 Diabetes Page 2 of 65 Abstract The molecular program underlying infrequent replication of pancreatic beta- cells remains largely inaccessible. Using transgenic mice expressing GFP in cycling cells we sorted live, replicating beta-cells and determined their transcriptome. Replicating beta-cells upregulate hundreds of proliferation- related genes, along with many novel putative cell cycle components. Strikingly, genes involved in beta-cell functions, namely glucose sensing and insulin secretion were repressed. Further studies using single molecule RNA in situ hybridization revealed that in fact, replicating beta-cells double the amount of RNA for most genes, but this upregulation excludes genes involved in beta-cell function. -

Downregulation of SNRPG Induces Cell Cycle Arrest and Sensitizes Human Glioblastoma Cells to Temozolomide by Targeting Myc Through a P53-Dependent Signaling Pathway
Cancer Biol Med 2020. doi: 10.20892/j.issn.2095-3941.2019.0164 ORIGINAL ARTICLE Downregulation of SNRPG induces cell cycle arrest and sensitizes human glioblastoma cells to temozolomide by targeting Myc through a p53-dependent signaling pathway Yulong Lan1,2*, Jiacheng Lou2*, Jiliang Hu1, Zhikuan Yu1, Wen Lyu1, Bo Zhang1,2 1Department of Neurosurgery, Shenzhen People’s Hospital, Second Clinical Medical College of Jinan University, The First Affiliated Hospital of Southern University of Science and Technology, Shenzhen 518020, China;2 Department of Neurosurgery, The Second Affiliated Hospital of Dalian Medical University, Dalian 116023, China ABSTRACT Objective: Temozolomide (TMZ) is commonly used for glioblastoma multiforme (GBM) chemotherapy. However, drug resistance limits its therapeutic effect in GBM treatment. RNA-binding proteins (RBPs) have vital roles in posttranscriptional events. While disturbance of RBP-RNA network activity is potentially associated with cancer development, the precise mechanisms are not fully known. The SNRPG gene, encoding small nuclear ribonucleoprotein polypeptide G, was recently found to be related to cancer incidence, but its exact function has yet to be elucidated. Methods: SNRPG knockdown was achieved via short hairpin RNAs. Gene expression profiling and Western blot analyses were used to identify potential glioma cell growth signaling pathways affected by SNRPG. Xenograft tumors were examined to determine the carcinogenic effects of SNRPG on glioma tissues. Results: The SNRPG-mediated inhibitory effect on glioma cells might be due to the targeted prevention of Myc and p53. In addition, the effects of SNRPG loss on p53 levels and cell cycle progression were found to be Myc-dependent. Furthermore, SNRPG was increased in TMZ-resistant GBM cells, and downregulation of SNRPG potentially sensitized resistant cells to TMZ, suggesting that SNRPG deficiency decreases the chemoresistance of GBM cells to TMZ via the p53 signaling pathway. -

Genomic Analysis of Estrogen Cascade Reveals Histone Variant H2A.Z Associated with Breast Cancer Progression
Molecular Systems Biology 4; Article number 188; doi:10.1038/msb.2008.25 Citation: Molecular Systems Biology 4:188 & 2008 EMBO and Nature Publishing Group All rights reserved 1744-4292/08 www.molecularsystemsbiology.com Genomic analysis of estrogen cascade reveals histone variant H2A.Z associated with breast cancer progression Sujun Hua1,2,3,9, Caleb B Kallen4,9, Ruby Dhar1,2, Maria T Baquero5, Christopher E Mason6, Beth A Russell1,2,6, Parantu K Shah1,2, Jiang Liu1,2, Andrey Khramtsov7, Maria S Tretiakova8, Thomas N Krausz8, Olufunmilayo I Olopade7, David L Rimm5 and Kevin P White1,2,* 1 Joint Institute for Genomics and Systems Biology, The University of Chicago and Argonne National Laboratory, Chicago, IL, USA, 2 Department of Human Genetics, The University of Chicago, Chicago, IL, USA, 3 Interdepartmental Program in Computational Biology and Bioinformatics, Yale University, New Haven, CT, USA, 4 Department of Gynecology and Obstetrics, Emory University, Atlanta, GA, USA, 5 Department of Pathology, Yale University, New Haven, CT, USA, 6 Department of Genetics, Yale University, New Haven, CT, USA, 7 Center for Clinical Cancer Genetics, University of Chicago Medical Center, Chicago, IL, USA and 8 Department of Pathology, The University of Chicago Hospitals, Chicago, IL, USA 9 These authors contributed equally to this work * Corresponding author. Department of Human Genetics, The University of Chicago, 1639 Pierce Drive, WMB 4211, Chicago, IL 60637, USA. Tel.: þ 1 773 834 8259; Fax: þ 1 773 834 0505; E-mail: [email protected] Received 7.9.07; accepted 13.3.08 We demonstrate an integrated approach to the study of a transcriptional regulatory cascade involved in the progression of breast cancer and we identify a protein associated with disease progression.