CHAF1A–PCNA Interaction Promotes Cervical Cancer Progression Via the PI3K/AKT/Foxo1 Signaling Pathway
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Maintenance of Self-Renewal Ability of Mouse Embryonic Stem Cells in The
MaintenanceBlackwellMalden,GTCGenes1365-2443©?117OriginalDnmt1/3a/3bA 2006 TsumuraBlackwell to USA ArticleCells Publishing et Publishing al. triple knockoutInc Ltd ES cells of self-renewal ability of mouse embryonic stem cells in the absence of DNA methyltransferases Dnmt1, Dnmt3a and Dnmt3b Akiko Tsumura1,4, Tomohiro Hayakawa2, Yuichi Kumaki3,6, Shin-ichiro Takebayashi1, Morito Sakaue1, Chisa Matsuoka1, Kunitada Shimotohno4, Fuyuki Ishikawa5, En Li7, Hiroki R. Ueda3, Jun-ichi Nakayama2 and Masaki Okano1,* 1Laboratory for Mammalian Epigenetic Studies, 2Laboratory for Chromatin Dynamics, and 3Laboratory for Systems Biology, Center for Developmental Biology, RIKEN, 2-2-3 Minatojima-Minamimachi, Chuo-ku, Kobe 650-0047, Japan 4Department of Viral Oncology, Institute for Virus Research, Kyoto University, 53 Kawaharacho, Shogoin, Sakyo-ku, Kyoto 606-8501, Japan 5Department of Gene Mechanisms, Graduate School of Biostudies, Kyoto University, Kitashirakawa-Oiwake-cho, Sakyo-ku, Kyoto 606-8502, Japan 6INTEC Web and Genome Informatics Corp., 1-3-3 Shinsuna, Koto-ku, Tokyo 136-8637, Japan 7Epigenetics Program, Novartis Institute for Biomedical Research, 250 Massachusetts Ave., Cambridge, MA 02139, USA DNA methyltransferases Dnmt1, Dnmt3a and Dnmt3b cooperatively regulate cytosine methylation in CpG dinucleotides in mammalian genomes, providing an epigenetic basis for gene silencing and maintenance of genome integrity. Proper CpG methylation is required for the normal growth of various somatic cell types, indicating its essential role in the basic cellular function of mammalian cells. Previous studies using Dnmt1–/– or Dnmt3a–/–Dnmt3b–/– ES cells, however, have shown that undifferentiated embryonic stem (ES) cells can tolerate hypomethylation for their proliferation. In an attempt to investigate the effects of the complete loss of CpG DNA methyltransferase function, we established mouse ES cells lacking all three of these enzymes by gene targeting. -

Regulation of Oxidized Base Damage Repair by Chromatin Assembly Factor 1 Subunit a Chunying Yang1,*,†, Shiladitya Sengupta1,2,*,†, Pavana M
Published online 27 October 2016 Nucleic Acids Research, 2017, Vol. 45, No. 2 739–748 doi: 10.1093/nar/gkw1024 Regulation of oxidized base damage repair by chromatin assembly factor 1 subunit A Chunying Yang1,*,†, Shiladitya Sengupta1,2,*,†, Pavana M. Hegde1,JoyMitra1, Shuai Jiang3, Brooke Holey3, Altaf H. Sarker3, Miaw-Sheue Tsai3, Muralidhar L. Hegde1,2,4 and Sankar Mitra1,2,* 1Department of Radiation Oncology, Houston Methodist Research Institute, Houston, TX 77030, USA, 2Weill Cornell Medical College, Cornell University, New York, NY 10065, USA, 3Biological Systems and Engineering Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA and 4Houston Methodist Neurological Institute, Houston, TX 77030, USA Received March 23, 2016; Revised October 13, 2016; Editorial Decision October 17, 2016; Accepted October 19, 2016 ABSTRACT INTRODUCTION Reactive oxygen species (ROS), generated both en- ROS, continuously generated in mammalian cells both en- dogenously and in response to exogenous stress, in- dogenously and by environmental genotoxicants, induce duce point mutations by mis-replication of oxidized various genomic lesions, including oxidized bases, abasic bases and other lesions in the genome. Repair of (AP) sites and single-strand breaks (SSBs). If unrepaired or these lesions via base excision repair (BER) pathway mis-repaired, DNA lesions would cause mutations which may lead to cytotoxicity and cell death and also carcino- maintains genomic fidelity. Regulation of the BER genic transformation (1). The base excision repair (BER) pathway for mutagenic oxidized bases, initiated by pathway, responsible for repair of oxidized base lesions NEIL1 and other DNA glycosylases at the chromatin which contribute to drug/radiation sensitivity is highly con- level remains unexplored. -

Supplementary Data
SUPPLEMENTARY DATA A cyclin D1-dependent transcriptional program predicts clinical outcome in mantle cell lymphoma Santiago Demajo et al. 1 SUPPLEMENTARY DATA INDEX Supplementary Methods p. 3 Supplementary References p. 8 Supplementary Tables (S1 to S5) p. 9 Supplementary Figures (S1 to S15) p. 17 2 SUPPLEMENTARY METHODS Western blot, immunoprecipitation, and qRT-PCR Western blot (WB) analysis was performed as previously described (1), using cyclin D1 (Santa Cruz Biotechnology, sc-753, RRID:AB_2070433) and tubulin (Sigma-Aldrich, T5168, RRID:AB_477579) antibodies. Co-immunoprecipitation assays were performed as described before (2), using cyclin D1 antibody (Santa Cruz Biotechnology, sc-8396, RRID:AB_627344) or control IgG (Santa Cruz Biotechnology, sc-2025, RRID:AB_737182) followed by protein G- magnetic beads (Invitrogen) incubation and elution with Glycine 100mM pH=2.5. Co-IP experiments were performed within five weeks after cell thawing. Cyclin D1 (Santa Cruz Biotechnology, sc-753), E2F4 (Bethyl, A302-134A, RRID:AB_1720353), FOXM1 (Santa Cruz Biotechnology, sc-502, RRID:AB_631523), and CBP (Santa Cruz Biotechnology, sc-7300, RRID:AB_626817) antibodies were used for WB detection. In figure 1A and supplementary figure S2A, the same blot was probed with cyclin D1 and tubulin antibodies by cutting the membrane. In figure 2H, cyclin D1 and CBP blots correspond to the same membrane while E2F4 and FOXM1 blots correspond to an independent membrane. Image acquisition was performed with ImageQuant LAS 4000 mini (GE Healthcare). Image processing and quantification were performed with Multi Gauge software (Fujifilm). For qRT-PCR analysis, cDNA was generated from 1 µg RNA with qScript cDNA Synthesis kit (Quantabio). qRT–PCR reaction was performed using SYBR green (Roche). -

Histone Chaperone CHAF1A Inhibits Differentiation and Promotes Aggressive Neuroblastoma
Published OnlineFirst December 12, 2013; DOI: 10.1158/0008-5472.CAN-13-1315 Cancer Molecular and Cellular Pathobiology Research Histone Chaperone CHAF1A Inhibits Differentiation and Promotes Aggressive Neuroblastoma Eveline Barbieri1, Katleen De Preter3, Mario Capasso4, Zaowen Chen1, Danielle M. Hsu2, Gian Paolo Tonini5, Steve Lefever3, John Hicks1, Rogier Versteeg7, Andrea Pession6, Frank Speleman3, Eugene S. Kim2, and Jason M. Shohet1 Abstract Neuroblastoma arises from the embryonal neural crest secondary to a block in differentiation. Long-term patient survival correlates inversely with the extent of differentiation, and treatment with retinoic acid or other prodifferentiation agents improves survival modestly. In this study, we show the histone chaperone and epigenetic regulator CHAF1A functions in maintaining the highly dedifferentiated state of this aggressive malignancy. CHAF1A is a subunit of the chromatin modifier chromatin assembly factor 1 and it regulates H3K9 trimethylation of key target genes regulating proliferation, survival, and differentiation. Elevated CHAF1A expression strongly correlated with poor prognosis. Conversely, CHAF1A loss-of-function was sufficient to drive neuronal differentiation in vitro and in vivo. Transcriptome analysis of cells lacking CHAF1A revealed repression of oncogenic signaling pathways and a normalization of glycolytic metabolism. Our findings demonstrate that CHAF1A restricts neural crest differentiation and contributes to the pathogenesis of high-risk neuroblastoma. Cancer Res; 74(3); 1–10. -

Specificity, Propagation, and Memory of Pericentric Heterochromatin
Article Specificity, propagation, and memory of pericentric heterochromatin Katharina Müller-Ott1, Fabian Erdel1, Anna Matveeva2, Jan-Philipp Mallm1, Anne Rademacher1, Matthias Hahn3, Caroline Bauer1, Qin Zhang2, Sabine Kaltofen1,†, Gunnar Schotta3, Thomas Höfer2 & Karsten Rippe1,* Abstract (Berger et al, 2009). These chromatin signals in turn recruit archi- tectural chromatin components or chromatin remodeling factors in The cell establishes heritable patterns of active and silenced chro- a highly dynamic manner and regulate genome access (McBryant matin via interacting factors that set, remove, and read epigenetic et al, 2006; Taverna et al, 2007; Campos & Reinberg, 2009; Clapier marks. To understand how the underlying networks operate, we & Cairns, 2009; Erdel et al, 2011a). On a global scale, the concerted have dissected transcriptional silencing in pericentric heterochro- and targeted activity of these networks results in the formation of matin (PCH) of mouse fibroblasts. We assembled a quantitative the denser, transcriptionally repressed heterochromatin state and map for the abundance and interactions of 16 factors related to the more open and biologically active euchromatin, which can be PCH in living cells and found that stably bound complexes of the distinguished at the resolution of the light microscope (Grewal & histone methyltransferase SUV39H1/2 demarcate the PCH state. Jia, 2007; Eissenberg & Reuter, 2009). A prototypic example for a From the experimental data, we developed a predictive mathe- constitutive heterochromatin domain is pericentric heterochromatin matical model that explains how chromatin-bound SUV39H1/2 (PCH) in mouse cells (Probst & Almouzni, 2008). It is characterized complexes act as nucleation sites and propagate a spatially by its high content of repetitive major satellite repeats and repres- confined PCH domain with elevated histone H3 lysine 9 trimethyla- sive epigenetic marks such as 5-methylcytosine (5meC) the binding tion levels via chromatin dynamics. -

Genome-Wide DNA Methylation Analysis Reveals Molecular Subtypes of Pancreatic Cancer
www.impactjournals.com/oncotarget/ Oncotarget, 2017, Vol. 8, (No. 17), pp: 28990-29012 Research Paper Genome-wide DNA methylation analysis reveals molecular subtypes of pancreatic cancer Nitish Kumar Mishra1 and Chittibabu Guda1,2,3,4 1Department of Genetics, Cell Biology and Anatomy, University of Nebraska Medical Center, Omaha, NE, 68198, USA 2Bioinformatics and Systems Biology Core, University of Nebraska Medical Center, Omaha, NE, 68198, USA 3Department of Biochemistry and Molecular Biology, University of Nebraska Medical Center, Omaha, NE, 68198, USA 4Fred and Pamela Buffet Cancer Center, University of Nebraska Medical Center, Omaha, NE, 68198, USA Correspondence to: Chittibabu Guda, email: [email protected] Keywords: TCGA, pancreatic cancer, differential methylation, integrative analysis, molecular subtypes Received: October 20, 2016 Accepted: February 12, 2017 Published: March 07, 2017 Copyright: Mishra et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC-BY), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Pancreatic cancer (PC) is the fourth leading cause of cancer deaths in the United States with a five-year patient survival rate of only 6%. Early detection and treatment of this disease is hampered due to lack of reliable diagnostic and prognostic markers. Recent studies have shown that dynamic changes in the global DNA methylation and gene expression patterns play key roles in the PC development; hence, provide valuable insights for better understanding the initiation and progression of PC. In the current study, we used DNA methylation, gene expression, copy number, mutational and clinical data from pancreatic patients. -

H19 Lncrna Controls Gene Expression of the Imprinted Gene Network by Recruiting MBD1
H19 lncRNA controls gene expression of the Imprinted Gene Network by recruiting MBD1 Paul Monniera,b, Clémence Martineta, Julien Pontisc, Irina Stanchevad, Slimane Ait-Si-Alic, and Luisa Dandoloa,1 aGenetics and Development Department, Institut National de la Santé et de la Recherche Médicale U1016, Centre National de la Recherche Scientifique (CNRS) Unité Mixte de Recherche (UMR) 8104, University Paris Descartes, Institut Cochin, Paris 75014, France; bUniversity Paris Pierre and Marie Curie, Paris 75005, France; cUniversity Paris Diderot, Sorbonne Paris Cité, Laboratoire Epigénétique et Destin Cellulaire, CNRS UMR 7216, Paris 75013, France; and dWellcome Trust Centre for Cell Biology, University of Edinburgh, Edinburgh EH9 3JR, United Kingdom Edited by Marisa Bartolomei, University of Pennsylvania, Philadelphia, PA, and accepted by the Editorial Board November 11, 2013 (received for review May 30, 2013) The H19 gene controls the expression of several genes within the this control is transcriptional or posttranscriptional and whether Imprinted Gene Network (IGN), involved in growth control of the these nine targets are direct or indirect targets remain elusive. embryo. However, the underlying mechanisms of this control re- Several lncRNAs interact with chromatin-modifying com- main elusive. Here, we identified the methyl-CpG–binding domain plexes and appear to exert a transcriptional control by targeting fi protein 1 MBD1 as a physical and functional partner of the H19 local chromatin modi cations at discrete genomic regions (8, 9). long noncoding RNA (lncRNA). The H19 lncRNA–MBD1 complex is In the case of imprinted clusters, the DMRs controlling the ex- fi pression of imprinted genes exhibit parent-of-origin epigenetic required for the control of ve genes of the IGN. -

Differential Expression Profile Analysis of DNA Damage Repair Genes in CD133+/CD133‑ Colorectal Cancer Cells
ONCOLOGY LETTERS 14: 2359-2368, 2017 Differential expression profile analysis of DNA damage repair genes in CD133+/CD133‑ colorectal cancer cells YUHONG LU1*, XIN ZHOU2*, QINGLIANG ZENG2, DAISHUN LIU3 and CHANGWU YUE3 1College of Basic Medicine, Zunyi Medical University, Zunyi; 2Deparment of Gastroenterological Surgery, Affiliated Hospital of Zunyi Medical University, Zunyi;3 Zunyi Key Laboratory of Genetic Diagnosis and Targeted Drug Therapy, The First People's Hospital of Zunyi, Zunyi, Guizhou 563003, P.R. China Received July 20, 2015; Accepted January 6, 2017 DOI: 10.3892/ol.2017.6415 Abstract. The present study examined differential expression cells. By contrast, 6 genes were downregulated and none levels of DNA damage repair genes in COLO 205 colorectal were upregulated in the CD133+ cells compared with the cancer cells, with the aim of identifying novel biomarkers for COLO 205 cells. These findings suggest that CD133+ cells the molecular diagnosis and treatment of colorectal cancer. may possess the same DNA repair capacity as COLO 205 COLO 205-derived cell spheres were cultured in serum-free cells. Heterogeneity in the expression profile of DNA damage medium supplemented with cell factors, and CD133+/CD133- repair genes was observed in COLO 205 cells, and COLO cells were subsequently sorted using an indirect CD133 205-derived CD133- cells and CD133+ cells may therefore microbead kit. In vitro differentiation and tumorigenicity assays provide a reference for molecular diagnosis, therapeutic target in BABA/c nude mice were performed to determine whether selection and determination of the treatment and prognosis for the CD133+ cells also possessed stem cell characteristics, in colorectal cancer. -

MBD1 Antibody
BioVision 09/14 For research use only MBD1 Antibody ChIP assays were performed using U2OS cells and the antibody and ALTERNATE NAMES: CXXC3, PCM1, RFT, Methyl-CpG-Binding Domain 1 optimized PCR primer sets for qPCR. Beads only were used as a negative CATALOG #: 6828-25 control. The Fig shows the recovery, expressed as a % of input (the relative AMOUNT: 25 µg amount of IP DNA compared to input DNA after qPCR analysis). HOST/ISOTYPE: Rabbit IMMUNOGEN: MBD1 (Methyl-CpG-binding domain protein 1) synthetic peptide containing a sequence from the N-terminal conjugated to KLH FORM: Liquid FORMULATION: In PBS containing 0.05% azide and 0.05% ProClin 300. PURIFICATION: Affinity purified SPECIES REACTIVITY: Human. STORAGE CONDITIONS: Store at -20°C; for long storage, store at -80°C. Avoid multiple freeze-thaw cycles. To determine the titer, an ELISA was performed using a serial dilution of the DESCRIPTION: MBD1 is a transcriptional repressor that specifically binds to methylated antibody. The antigen used was a CpG dinucleotides in promoter sequences. MBD1 acts by recruiting a variety of histone peptide containing the histone deacetylases (HDAC’s) and chromatin remodelling factors. MBD1-dependent transcriptional modification of interest. By plotting the repression is mediated by ATF7IP through the recruitment of factors such as the histone absorbance against the antibody methyltransferase SETDB1. MBD1 probably forms a complex with SETDB1 and ATF7IP dilution the titer of the antibody was which couples DNA methylation to H3K9 trimethylation and represses transcription. estimated to be 1:20,000. APPLICATION: ChIP: 1.5 µg/ChIP, WB: 1:500, ELISA: 1:1000. -
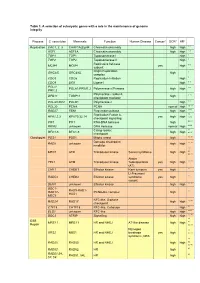
Table 1. a Selection of Eukaryotic Genes with a Role in the Maintenance of Genome Integrity
Table 1. A selection of eukaryotic genes with a role in the maintenance of genome integrity Process S. cerevisiae Mammals Function Human Disease Cancer* GCR* HR* Replication CAC1, 2, 3 CHAF1A,B,p48 Chromatin assembly high high 1, 2 1, 2 ASF1 ASF1A Chromatin assembly high high 3 TOP1 TOP1 Topoisomerase I high 3 TOP2 TOP2 Topoisomerase II high Replicative helicase 4, 5 MCM4 MCM4 yes high subunit Origin Replication 6 ORC3/5 ORC3/6L high complex 7 CDC6 CDC6 Replication Initiation high 7, 8 CDC9 LIG1 Ligase I high POL1/ 7-10 POLA1/PRIM1,2 Polymerase α/Primase high high PRI1,2 Polymerase ε subunit, 6, 11 DPB11 TOBP11 high checkpoint mediator POL3/CDC2 POLD1 Polymerase δ high 7, 8 POL30 PCNA PCNA normal high 12, 13 RAD27 FEN1 Flap endonuclease high high 14-16 Replication Factor A, 14, RFA1,2,3 RPA70,32,14 yes high high 17-19 checkpoint signalling PIF1 PIF1 RNA-DNA helicase high 20, 21 RRM3 unknown DNA Helicase normal high 22-26 Clamp loader, 11, RFC1-5 RFC1-5 high high 20, 27 checkpoint Checkpoint PDS1 PDS1 Mitotic arrest high 11, 28 Damage checkpoint 11, 29 RAD9 unknown high high mediator 11, MEC1 ATR Transducer kinase Seckel syndrome high high 28, 30, 31 Ataxia TEL1 ATM Transducer kinase Telangiectasia yes high high 11, 28 (AT) CHK1 CHEK1 Effector kinase Rare tumours yes high 11 Li-Fraumeni RAD53 CHEK2 Effector kinase syndrome yes high 11 variant DUN1 unknown Effector kinase high high 11, 32 DDC1- RAD9-RAD1- 11 RAD17- PCNA-like complex high HUS1 MEC3 RFC-like, S-phase 11, 33 RAD24 RAD17 high high checkponit 34 CTF18 CHTF18 RFC-like, Cohesion -

The Genetic Program of Pancreatic Beta-Cell Replication in Vivo
Page 1 of 65 Diabetes The genetic program of pancreatic beta-cell replication in vivo Agnes Klochendler1, Inbal Caspi2, Noa Corem1, Maya Moran3, Oriel Friedlich1, Sharona Elgavish4, Yuval Nevo4, Aharon Helman1, Benjamin Glaser5, Amir Eden3, Shalev Itzkovitz2, Yuval Dor1,* 1Department of Developmental Biology and Cancer Research, The Institute for Medical Research Israel-Canada, The Hebrew University-Hadassah Medical School, Jerusalem 91120, Israel 2Department of Molecular Cell Biology, Weizmann Institute of Science, Rehovot, Israel. 3Department of Cell and Developmental Biology, The Silberman Institute of Life Sciences, The Hebrew University of Jerusalem, Jerusalem 91904, Israel 4Info-CORE, Bioinformatics Unit of the I-CORE Computation Center, The Hebrew University and Hadassah, The Institute for Medical Research Israel- Canada, The Hebrew University-Hadassah Medical School, Jerusalem 91120, Israel 5Endocrinology and Metabolism Service, Department of Internal Medicine, Hadassah-Hebrew University Medical Center, Jerusalem 91120, Israel *Correspondence: [email protected] Running title: The genetic program of pancreatic β-cell replication 1 Diabetes Publish Ahead of Print, published online March 18, 2016 Diabetes Page 2 of 65 Abstract The molecular program underlying infrequent replication of pancreatic beta- cells remains largely inaccessible. Using transgenic mice expressing GFP in cycling cells we sorted live, replicating beta-cells and determined their transcriptome. Replicating beta-cells upregulate hundreds of proliferation- related genes, along with many novel putative cell cycle components. Strikingly, genes involved in beta-cell functions, namely glucose sensing and insulin secretion were repressed. Further studies using single molecule RNA in situ hybridization revealed that in fact, replicating beta-cells double the amount of RNA for most genes, but this upregulation excludes genes involved in beta-cell function. -

Mice Lacking Methyl-Cpg Binding Protein 1 Have Deficits in Adult Neurogenesis and Hippocampal Function
Mice lacking methyl-CpG binding protein 1 have deficits in adult neurogenesis and hippocampal function Xinyu Zhao*, Tetsuya Ueba*†, Brian R. Christie‡, Basam Barkho*, Michael J. McConnell§, Kinichi Nakashima*, Edward S. Lein*, Brennan D. Eadie‡, Andrew R. Willhoite*, Alysson R. Muotri*, Robert G. Summers*, Jerold Chun§, Kuo-Fen Lee¶, and Fred H. Gage*ʈ *Laboratory of Genetics and ¶Peptide Biology Laboratory, The Salk Institute for Biological Studies, La Jolla, CA 92037; §Department of Pharmacology, University of California, 9500 Gilman Drive, La Jolla, CA 92093; and ‡Department of Psychology, University of British Columbia, 2136 West Mall, Vancouver, BC, Canada V6T 1Z4 Communicated by Inder M. Verma, The Salk Institute for Biological Studies, San Diego, CA, April 2, 2003 (received for review March 7, 2003) DNA methylation-mediated epigenetic regulation plays critical roles in vivo function of MBD1 is still unclear. Because MBD1 has no in regulating mammalian gene expression, but its role in normal brain sequence specificity, except for methylated CpG, its in vivo target function is not clear. Methyl-CpG binding protein 1 (MBD1), a member genes are not known. of the methylated DNA-binding protein family, has been shown to Maintaining normal DNA methylation levels is critical during bind methylated gene promoters and facilitate transcriptional repres- mammalian development, as demonstrated by the embryonic Ϫ Ϫ sion in vitro. Here we report the generation and analysis of MBD1؊/؊ lethality of Dnmt1 / mice (17). Mutation of the de novo DNA -mice. MBD1؊/؊ mice had no detectable developmental defects and methyltransferase 1-36 (Dnmt3b) causes immunodeficiency, cen -appeared healthy throughout life. However, we found that MBD1؊/؊ tromeric instability, and facial anomalies (ICF) syndrome in hu neural stem cells exhibited reduced neuronal differentiation and mans, with characteristics of genomic instability (18).