NRF1 and NRF3 Complementarily Maintain a Basal Proteasome Activity in Cancer Cells Through CPEB3-Mediated Translational Repressi
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Subterranean Mammals Show Convergent Regression in Ocular Genes and Enhancers, Along with Adaptation to Tunneling
RESEARCH ARTICLE Subterranean mammals show convergent regression in ocular genes and enhancers, along with adaptation to tunneling Raghavendran Partha1, Bharesh K Chauhan2,3, Zelia Ferreira1, Joseph D Robinson4, Kira Lathrop2,3, Ken K Nischal2,3, Maria Chikina1*, Nathan L Clark1* 1Department of Computational and Systems Biology, University of Pittsburgh, Pittsburgh, United States; 2UPMC Eye Center, Children’s Hospital of Pittsburgh, Pittsburgh, United States; 3Department of Ophthalmology, University of Pittsburgh School of Medicine, Pittsburgh, United States; 4Department of Molecular and Cell Biology, University of California, Berkeley, United States Abstract The underground environment imposes unique demands on life that have led subterranean species to evolve specialized traits, many of which evolved convergently. We studied convergence in evolutionary rate in subterranean mammals in order to associate phenotypic evolution with specific genetic regions. We identified a strong excess of vision- and skin-related genes that changed at accelerated rates in the subterranean environment due to relaxed constraint and adaptive evolution. We also demonstrate that ocular-specific transcriptional enhancers were convergently accelerated, whereas enhancers active outside the eye were not. Furthermore, several uncharacterized genes and regulatory sequences demonstrated convergence and thus constitute novel candidate sequences for congenital ocular disorders. The strong evidence of convergence in these species indicates that evolution in this environment is recurrent and predictable and can be used to gain insights into phenotype–genotype relationships. DOI: https://doi.org/10.7554/eLife.25884.001 *For correspondence: [email protected] (MC); [email protected] (NLC) Competing interests: The Introduction authors declare that no The subterranean habitat has been colonized by numerous animal species for its shelter and unique competing interests exist. -

The Ubiquitin Proteasome System Is Required for Cell Proliferation of The
RESEARCH ARTICLE 3257 Development 137, 3257-3268 (2010) doi:10.1242/dev.053124 © 2010. Published by The Company of Biologists Ltd The ubiquitin proteasome system is required for cell proliferation of the lens epithelium and for differentiation of lens fiber cells in zebrafish Fumiyasu Imai1, Asuka Yoshizawa1, Noriko Fujimori-Tonou2, Koichi Kawakami3 and Ichiro Masai1,* SUMMARY In the developing vertebrate lens, epithelial cells differentiate into fiber cells, which are elongated and flat in shape and form a multilayered lens fiber core. In this study, we identified the zebrafish volvox (vov) mutant, which shows defects in lens fiber differentiation. In the vov mutant, lens epithelial cells fail to proliferate properly. Furthermore, differentiating lens fiber cells do not fully elongate, and the shape and position of lens fiber nuclei are affected. We found that the vov mutant gene encodes Psmd6, the subunit of the 26S proteasome. The proteasome regulates diverse cellular functions by degrading polyubiquitylated proteins. Polyubiquitylated proteins accumulate in the vov mutant. Furthermore, polyubiquitylation is active in nuclei of differentiating lens fiber cells, suggesting roles of the proteasome in lens fiber differentiation. We found that an E3 ubiquitin ligase anaphase-promoting complex/cyclosome (APC/C) is involved in lens defects in the vov mutant. These data suggest that the ubiquitin proteasome system is required for cell proliferation of lens epithelium and for the differentiation of lens fiber cells in zebrafish. KEY WORDS: Ubiquitin, Proteasome, Lens, Zebrafish, Psmd6, APC/C INTRODUCTION acid DNase (DLAD; Dnase2b). However, it is unclear how lens During development, the lens placode delaminates from the morphogenesis and organelle loss are related to these transcription epidermal ectoderm and forms the lens vesicle. -

Genetic Analysis of Complex Traits in the Emerging Collaborative Cross
Downloaded from genome.cshlp.org on October 5, 2021 - Published by Cold Spring Harbor Laboratory Press Research Genetic analysis of complex traits in the emerging Collaborative Cross David L. Aylor,1 William Valdar,1,13 Wendy Foulds-Mathes,1,13 Ryan J. Buus,1,13 Ricardo A. Verdugo,2,13 Ralph S. Baric,3,4 Martin T. Ferris,1 Jeff A. Frelinger,4 Mark Heise,1 Matt B. Frieman,4 Lisa E. Gralinski,4 Timothy A. Bell,1 John D. Didion,1 Kunjie Hua,1 Derrick L. Nehrenberg,1 Christine L. Powell,1 Jill Steigerwalt,5 Yuying Xie,1 Samir N.P. Kelada,6 Francis S. Collins,6 Ivana V. Yang,7 David A. Schwartz,7 Lisa A. Branstetter,8 Elissa J. Chesler,2 Darla R. Miller,1 Jason Spence,1 Eric Yi Liu,9 Leonard McMillan,9 Abhishek Sarkar,9 Jeremy Wang,9 Wei Wang,9 Qi Zhang,9 Karl W. Broman,10 Ron Korstanje,2 Caroline Durrant,11 Richard Mott,11 Fuad A. Iraqi,12 Daniel Pomp,1,14 David Threadgill,5,14 Fernando Pardo-Manuel de Villena,1,14 and Gary A. Churchill2,14 1Department of Genetics, University of North Carolina–Chapel Hill, Chapel Hill, North Carolina 27599, USA; 2The Jackson Laboratory, Bar Harbor, Maine 04609, USA; 3Department of Epidemiology, University of North Carolina–Chapel Hill, Chapel Hill, North Carolina 27599, USA; 4Department of Microbiology and Immunology, University of North Carolina–Chapel Hill, Chapel Hill, North Carolina 27599, USA; 5Department of Genetics, North Carolina State University, Raleigh, North Carolina 27695, USA; 6Genome Technology Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland -

15141 POMP (D2X9S) Rabbit Mab
Revision 1 C 0 2 - t POMP (D2X9S) Rabbit mAb a e r o t S Orders: 877-616-CELL (2355) [email protected] 1 Support: 877-678-TECH (8324) 4 1 Web: [email protected] 5 www.cellsignal.com 1 # 3 Trask Lane Danvers Massachusetts 01923 USA For Research Use Only. Not For Use In Diagnostic Procedures. Applications: Reactivity: Sensitivity: MW (kDa): Source/Isotype: UniProt ID: Entrez-Gene Id: WB, IP H M R Mk Endogenous 16 Rabbit IgG Q9Y244 51371 Product Usage Information immunoproteasomes, and that the assembly focal point resides at the endoplasmic reticulum (6-8). A single nucleotide deletion in the 5' UTR of POMP results in altered Application Dilution epidermal POMP distribution and the autosomal recessive skin disorder known as KLICK syndrome (9). Western Blotting 1:1000 1. Finley, D. (2009) Annu Rev Biochem 78, 477-513. Immunoprecipitation 1:200 2. Lee, M.J. et al. (2011) Mol Cell Proteomics 10, R110.003871. 3. Griffin, T.A. et al. (2000) Mol Cell Biol Res Commun 3, 212-7. 4. Witt, E. et al. (2000) J Mol Biol 301, 1-9. Storage 5. Ramos, P.C. et al. (1998) Cell 92, 489-99. Supplied in 10 mM sodium HEPES (pH 7.5), 150 mM NaCl, 100 µg/ml BSA, 50% 6. Fricke, B. et al. (2007) EMBO Rep 8, 1170-5. glycerol and less than 0.02% sodium azide. Store at –20°C. Do not aliquot the antibody. 7. Heink, S. et al. (2005) Proc Natl Acad Sci U S A 102, 9241-6. 8. Heink, S. -
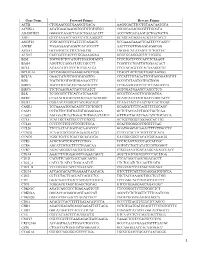
Gene Name Forward Primer Reverse Primer ACTB
Gene Name Forward Primer Reverse Primer ACTB CTGGAACGGTGAAGGTGACA AAGGGACTTCCTGTAACAATGCA ACVRL1 ACATGAAGAAGGTGGTGTGTGTGG CGGGCAGAGGGGTTTGGGTA ADAMDEC1 GGGGCCAGACTACACTGAAACATT ACCCGTCACAAGTACTGATGCTG AHI1 GTCCAAAACTACCCCATCAAGGCT GCAGCACAGGAACGTATCACCT ANGPT2 TGGCAGCGTTGATTTTCAGAGG GCGAAACAAACTCATTTCCCAGCC ANPEP TGAAGAAGCAGGTCACACCCCT AACTCCGTTGGAGCAGGCGG APOA1 GCCGTGCTCTTCCTGACGG TGGGACACATAGTCTCTGCCGC ATXN7 CACCGCCCACTCTGGAAAAGAA GGGTGCAGGGCTTCTTGGTG B2M TGCTGTCTCCATGTTTGATGTATCT TCTCTGCTCCCCACCTCTAAGT BAG4 AGGTTCCAGGATATCCGCCTT TCGGTCCTGATTGTGGAACACT BCL2 ACAACATCGCCCTGTGGATGA CCGTACAGTTCCACAAAGGCAT BCL2L14 GCTCAGGGTCAAAGGACGTTGG TCAGCTACTCGGTTGGCAATGG BCL7A GAACCATGTCGGGCAGGTCG CCCATTTGTAGATTCGTAGGGATGTGT BIN1 TGCTGTCGTGGTGGAGACCTTC GCCGTGTAGTCGTGCTGGG BIRC3 TGCTATCCACATCAGACAGCCC TCTGAATGGTCTTCTCCAGGTTCA BIRC5 TTCTCAAGGACCACCGCATCT AGTGGATGAAGCCAGCCTCG BLK TCGGGGTCTTCACCATCAAAGC GCGCTCCAGGTTGCGGATGA BTRC CCAAATGTGTCATTACCAACATGGGC GCAGCACATAGTGATTTGGCATCC BUB3 CGGAACATGGGTTACGTGCAGC CCAAATACTCAACTGCCACTCGGC CAGE1 TCCAAAATGCACAGTCTTCTGGCT GGAGGCTCTTCAGTTTTTGCAGC CASP1 CCTGTTCCTGTGATGTGGAGGAAA GCTCTACCATCTGGCTGCTCAA CASP3 AGCGAATCAATGGACTCTGGAATATCC GTTTGCTGCATCGACATCTGTACCA CCL5 TCATTGCTACTGCCCTCTGCG ACTGCTGGGTTGGAGCACTTG CCL18 CCCTCCTTGTCCTCGTCTGCA GCACTGGGGGCTGGTTTCAG CCL26 TTCCAATACAGCCACAAGCCCC GGATGGGTACAGACTTTCTTGCCTC CCND2 TCAAGTGCGTGCAGAAGGACAT CTTCGCACTTCTGTTCCTCACA CCND3 TGGCTGCTGTGATTGCACATGA GATGGCGGGTACATGGCAAAGG CCR3 ACGCTGCTCTGCTTCCTGG TCCTCAGTTCCCCACCATCGC CCR4 AGCATCGTGCTTCCTGAGCAA GGTGTCTGCTATATCCGTGGGGT CCR7 AGACAGGGGTAGTGCGAGGC -
![Myelogenous Leukemia 8;21 Translocation: Evidence That C-Mos Is Not Translocated [Somatic Cell Hybrids/C-Myc/Gene Mapping/Superoxide Dismutase (Soluble)] HARRY A](https://docslib.b-cdn.net/cover/6910/myelogenous-leukemia-8-21-translocation-evidence-that-c-mos-is-not-translocated-somatic-cell-hybrids-c-myc-gene-mapping-superoxide-dismutase-soluble-harry-a-2176910.webp)
Myelogenous Leukemia 8;21 Translocation: Evidence That C-Mos Is Not Translocated [Somatic Cell Hybrids/C-Myc/Gene Mapping/Superoxide Dismutase (Soluble)] HARRY A
Proc. Natl. Acad. Sci. USA Vol. 82, pp. 464-468, January 1985 Genetics Isolation and analysis of the 21q+ chromosome in the acute myelogenous leukemia 8;21 translocation: Evidence that c-mos is not translocated [somatic cell hybrids/c-myc/gene mapping/superoxide dismutase (soluble)] HARRY A. DRABKIN*t, MANUEL DIAZt, CYNTHIA M. BRADLEY*, MICHELLE M. LE BEAUS, JANET D. ROWLEYt, AND DAVID PATTERSON*§¶ *The Eleanor Roosevelt Institute for Cancer Research, 4200 East Ninth Avenue, B-129, and tThe Division of Medical Oncology and Departments of Medicine and ¶Biochemistry, Biophysics and Genetics, University of Colorado Health Sciences Center, 4200 East Ninth Avenue, Denver, CO 80262; and tSection of Hematology-Oncology, Department of Medicine, Pritzker School of Medicine, The University of Chicago, Chicago, IL 60637 Contributed by Janet D. Rowley, September 4, 1984 ABSTRACT Acute myelogenous leukemia (AML), sub- light chain genes are translocated to the 3' region of c-myc group M2, is associated with a nonrandom chromosomal (4-7). translocation, t(8;21)(q22,q22). The oncogene c-mos also has Somatic cell genetic approaches have been used also to been localized to the q22 band on chromosome 8. There is also show that, in Philadelphia (Ph')-positive chronic myeloge- evidence that genes on chromosome 21 may be important in nous leukemia, c-abl oncogene sequences are translocated the development of leukemia. To determine whether the c-mos from chromosome 9 to the Ph' (22q-) chromosome (8). oncogene has been translocated in AML-M2 with this translo- Analysis of cells from two Ph'-positive patients has revealed cation and to isolate DNA sequences and genes from these two that the breakpoint in chromosome 9 is near c-abl (9). -

Downregulation of Carnitine Acyl-Carnitine Translocase by Mirnas
Page 1 of 288 Diabetes 1 Downregulation of Carnitine acyl-carnitine translocase by miRNAs 132 and 212 amplifies glucose-stimulated insulin secretion Mufaddal S. Soni1, Mary E. Rabaglia1, Sushant Bhatnagar1, Jin Shang2, Olga Ilkayeva3, Randall Mynatt4, Yun-Ping Zhou2, Eric E. Schadt6, Nancy A.Thornberry2, Deborah M. Muoio5, Mark P. Keller1 and Alan D. Attie1 From the 1Department of Biochemistry, University of Wisconsin, Madison, Wisconsin; 2Department of Metabolic Disorders-Diabetes, Merck Research Laboratories, Rahway, New Jersey; 3Sarah W. Stedman Nutrition and Metabolism Center, Duke Institute of Molecular Physiology, 5Departments of Medicine and Pharmacology and Cancer Biology, Durham, North Carolina. 4Pennington Biomedical Research Center, Louisiana State University system, Baton Rouge, Louisiana; 6Institute for Genomics and Multiscale Biology, Mount Sinai School of Medicine, New York, New York. Corresponding author Alan D. Attie, 543A Biochemistry Addition, 433 Babcock Drive, Department of Biochemistry, University of Wisconsin-Madison, Madison, Wisconsin, (608) 262-1372 (Ph), (608) 263-9608 (fax), [email protected]. Running Title: Fatty acyl-carnitines enhance insulin secretion Abstract word count: 163 Main text Word count: 3960 Number of tables: 0 Number of figures: 5 Diabetes Publish Ahead of Print, published online June 26, 2014 Diabetes Page 2 of 288 2 ABSTRACT We previously demonstrated that micro-RNAs 132 and 212 are differentially upregulated in response to obesity in two mouse strains that differ in their susceptibility to obesity-induced diabetes. Here we show the overexpression of micro-RNAs 132 and 212 enhances insulin secretion (IS) in response to glucose and other secretagogues including non-fuel stimuli. We determined that carnitine acyl-carnitine translocase (CACT, Slc25a20) is a direct target of these miRNAs. -

Downloaded Per Proteome Cohort Via the Web- Site Links of Table 1, Also Providing Information on the Deposited Spectral Datasets
www.nature.com/scientificreports OPEN Assessment of a complete and classifed platelet proteome from genome‑wide transcripts of human platelets and megakaryocytes covering platelet functions Jingnan Huang1,2*, Frauke Swieringa1,2,9, Fiorella A. Solari2,9, Isabella Provenzale1, Luigi Grassi3, Ilaria De Simone1, Constance C. F. M. J. Baaten1,4, Rachel Cavill5, Albert Sickmann2,6,7,9, Mattia Frontini3,8,9 & Johan W. M. Heemskerk1,9* Novel platelet and megakaryocyte transcriptome analysis allows prediction of the full or theoretical proteome of a representative human platelet. Here, we integrated the established platelet proteomes from six cohorts of healthy subjects, encompassing 5.2 k proteins, with two novel genome‑wide transcriptomes (57.8 k mRNAs). For 14.8 k protein‑coding transcripts, we assigned the proteins to 21 UniProt‑based classes, based on their preferential intracellular localization and presumed function. This classifed transcriptome‑proteome profle of platelets revealed: (i) Absence of 37.2 k genome‑ wide transcripts. (ii) High quantitative similarity of platelet and megakaryocyte transcriptomes (R = 0.75) for 14.8 k protein‑coding genes, but not for 3.8 k RNA genes or 1.9 k pseudogenes (R = 0.43–0.54), suggesting redistribution of mRNAs upon platelet shedding from megakaryocytes. (iii) Copy numbers of 3.5 k proteins that were restricted in size by the corresponding transcript levels (iv) Near complete coverage of identifed proteins in the relevant transcriptome (log2fpkm > 0.20) except for plasma‑derived secretory proteins, pointing to adhesion and uptake of such proteins. (v) Underrepresentation in the identifed proteome of nuclear‑related, membrane and signaling proteins, as well proteins with low‑level transcripts. -

Download.Cse.Ucsc.Edu/Goldenpath/ Knowngene Mm7/Database/ Refgene All Mrna Knowntoensembl
BMC Genomics BioMed Central Methodology article Open Access Comparison of gene coverage of mouse oligonucleotide microarray platforms Ricardo A Verdugo and Juan F Medrano* Address: Department of Animal Science, University of California Davis, One Shields Avenue, Davis, CA 95616-8521, USA Email: Ricardo A Verdugo - [email protected]; Juan F Medrano* - [email protected] * Corresponding author Published: 21 March 2006 Received: 24 September 2005 Accepted: 21 March 2006 BMC Genomics2006, 7:58 doi:10.1186/1471-2164-7-58 This article is available from: http://www.biomedcentral.com/1471-2164/7/58 © 2006Verdugo and Medrano; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: The increasing use of DNA microarrays for genetical genomics studies generates a need for platforms with complete coverage of the genome. We have compared the effective gene coverage in the mouse genome of different commercial and noncommercial oligonucleotide microarray platforms by performing an in-house gene annotation of probes. We only used information about probes that is available from vendors and followed a process that any researcher may take to find the gene targeted by a given probe. In order to make consistent comparisons between platforms, probes in each microarray were annotated with an Entrez Gene id and the chromosomal position for each gene was obtained from the UCSC Genome Browser Database. Gene coverage was estimated as the percentage of Entrez Genes with a unique position in the UCSC Genome database that is tested by a given microarray platform. -

Gene Mapping and Medical Genetics Human Chromosome 8
J Med Genet: first published as 10.1136/jmg.25.11.721 on 1 November 1988. Downloaded from Gene mapping and medical genetics Journal of Medical Genetics 1988, 25, 721-731 Human chromosome 8 STEPHEN WOOD From the Department of Medical Genetics, University of British Columbia, 6174 University Boulevard, Vancouver, British Columbia, Canada V6T IWS. SUMMARY The role of human chromosome 8 in genetic disease together with the current status of the genetic linkage map for this chromosome is reviewed. Both hereditary genetic disease attributed to mutant alleles at gene loci on chromosome 8 and neoplastic disease owing to somatic mutation, particularly chromosomal translocations, are discussed. Human chromosome 8 is perhaps best known for its In an era when complete sequencing of the human involvement in Burkitt's lymphoma and as the genome is being proposed, it is appropriate for location of the tissue plasminogen activator gene, medical geneticists to accept the challenge of defining by copyright. PLAT, which has been genetically engineered to the set of loci that have mutant alleles causing provide a natural fibrinolytic product for emergency hereditary disease. The fundamental genetic tool of use in cardiac disease. Since chromosome 8 repre- linkage mapping can now be applied, owing largely sents about 5% of the human genome, we may to progress in defining RFLP markers.3 4 This expect it to carry about 5% of human gene loci. This review will focus on genetic disease associated with would correspond to about 90 of the fully validated chromosome 8 loci and the status ofthe chromosome 8 phenotypes in the MIM7 catalogue.' The 27 genes linkage map. -

SF3B1-Mutated Chronic Lymphocytic Leukemia Shows Evidence Of
SF3B1-mutated chronic lymphocytic leukemia shows evidence of NOTCH1 pathway activation including CD20 downregulation by Federico Pozzo, Tamara Bittolo, Erika Tissino, Filippo Vit, Elena Vendramini, Luca Laurenti, Giovanni D'Arena, Jacopo Olivieri, Gabriele Pozzato, Francesco Zaja, Annalisa Chiarenza, Francesco Di Raimondo, Antonella Zucchetto, Riccardo Bomben, Francesca Maria Rossi, Giovanni Del Poeta, Michele Dal Bo, and Valter Gattei Haematologica 2020 [Epub ahead of print] Citation: Federico Pozzo, Tamara Bittolo, Erika Tissino, Filippo Vit, Elena Vendramini, Luca Laurenti, Giovanni D'Arena, Jacopo Olivieri, Gabriele Pozzato, Francesco Zaja, Annalisa Chiarenza, Francesco Di Raimondo, Antonella Zucchetto, Riccardo Bomben, Francesca Maria Rossi, Giovanni Del Poeta, Michele Dal Bo, and Valter Gattei SF3B1-mutated chronic lymphocytic leukemia shows evidence of NOTCH1 pathway activation including CD20 downregulation. Haematologica. 2020; 105:xxx doi:10.3324/haematol.2020.261891 Publisher's Disclaimer. E-publishing ahead of print is increasingly important for the rapid dissemination of science. Haematologica is, therefore, E-publishing PDF files of an early version of manuscripts that have completed a regular peer review and have been accepted for publication. E-publishing of this PDF file has been approved by the authors. After having E-published Ahead of Print, manuscripts will then undergo technical and English editing, typesetting, proof correction and be presented for the authors' final approval; the final version of the manuscript will -

Genetic Amplification of the NOTCH Modulator LNX2 Upregulates The
Cancer Tumor and Stem Cell Biology Research Genetic Amplification of the NOTCH Modulator LNX2 Upregulates the WNT/b-Catenin Pathway in Colorectal Cancer Jordi Camps1, Jason J. Pitt2, Georg Emons1,3, Amanda B. Hummon1, Chanelle M. Case1, Marian Grade1,3, Tamara L. Jones2, Quang T. Nguyen1, B. Michael Ghadimi3, Tim Beissbarth3, Michael J. Difilippantonio1, Natasha J. Caplen2, and Thomas Ried1 Abstract Chromosomal copy number alterations (aneuploidy) define the genomic landscape of most cancer cells, but identification of the oncogenic drivers behind these imbalances remains an unfinished task. In this study, we conducted a systematic analysis of colorectal carcinomas that integrated genomic copy number changes and gene expression profiles. This analysis revealed 44 highly overexpressed genes mapping to localized amplicons on chromosome 13, gains of which occur often in colorectal cancers (CRC). RNA interference (RNAi)–mediated silencing identified eight candidates whose loss-of-function reduced cell viability 20% or more in CRC cell lines. The functional space of the genes NUPL1, LNX2, POLR1D, POMP, SLC7A1, DIS3, KLF5, and GPR180 was established by global expression profiling after RNAi exposure. One candidate, LNX2, not previously known as an oncogene, was involved in regulating NOTCH signaling. Silencing LNX2 reduced NOTCH levels but also downregulated the transcription factor TCF7L2 and markedly reduced WNT signaling. LNX2 overexpression and chromosome 13 amplification therefore constitutively activates the WNT pathway, offering evidence of an aberrant NOTCH–WNT axis in CRC. Cancer Res; 73(6); 1–11. Ó2012 AACR. Introduction band or less) or they affect entire chromosome arms or Colorectal cancer (CRC) has an annual incidence of some chromosomes, most commonly resulting in low level gains or 150,000 new cases and a mortality of more than 50,000 in the losses.