CDK8 : a New Modulator for X-Chromosome Inactivation
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-
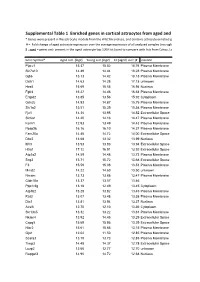
Supplemental Table 1 Enriched Genes in Cortical Astrocytes from Aged
Supplemental Table 1 Enriched genes in cortical astrocytes from aged and young-adult mice * Genes were present in the astrocyte module from the WGCNA analysis, and contains astrocyte enriched genes compared to microglia and oligodendrocytes # = Fold change of aged astrocyte expression over the average expression of all analyzed samples (microglia, astrocytes: young, old, with and without myelin contamination) $ ; aged = genes only present in the aged astrocyte top 1000 list (used to compare with lists from Cahoy, Lovatt, Doyle; see Fig. 4B), all = genes present in all astrocyte top 1000 lists Gene Symbol* Aged astr. (log2) Young astr.(log2) FC (aged/ aver.)# Location Ptprz1 15.37 15.02 18.76 Plasma Membrane Slc7a10 14.49 14.44 18.28 Plasma Membrane Gjb6 15.13 14.42 18.18 Plasma Membrane Dclk1 14.63 14.28 17.18 unknown Hes5 15.69 15.55 16.94 Nucleus Fgfr3 15.27 14.46 16.54 Plasma Membrane Entpd2 13.85 13.56 15.92 Cytoplasm Grin2c 14.93 14.87 15.75 Plasma Membrane Slc1a2 15.51 15.39 15.58 Plasma Membrane Fjx1 14.36 13.98 14.52 Extracellular Space Slc6a1 14.20 14.16 14.47 Plasma Membrane Kcnk1 12.93 13.49 14.43 Plasma Membrane Ppap2b 16.16 16.10 14.37 Plasma Membrane Fam20a 14.48 14.72 14.00 Extracellular Space Dbx2 13.68 13.32 13.99 Nucleus Itih3 13.93 13.93 13.94 Extracellular Space Htra1 17.12 16.91 13.92 Extracellular Space Atp1a2 14.59 14.48 13.73 Plasma Membrane Scg3 15.71 15.72 13.68 Extracellular Space F3 15.59 15.08 13.51 Plasma Membrane Mmd2 14.22 14.60 13.50 unknown Nrcam 13.73 13.88 13.47 Plasma Membrane Cldn10a 13.37 13.57 13.46 -

Transcriptome Analyses of Rhesus Monkey Pre-Implantation Embryos Reveal A
Downloaded from genome.cshlp.org on September 23, 2021 - Published by Cold Spring Harbor Laboratory Press Transcriptome analyses of rhesus monkey pre-implantation embryos reveal a reduced capacity for DNA double strand break (DSB) repair in primate oocytes and early embryos Xinyi Wang 1,3,4,5*, Denghui Liu 2,4*, Dajian He 1,3,4,5, Shengbao Suo 2,4, Xian Xia 2,4, Xiechao He1,3,6, Jing-Dong J. Han2#, Ping Zheng1,3,6# Running title: reduced DNA DSB repair in monkey early embryos Affiliations: 1 State Key Laboratory of Genetic Resources and Evolution, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, Yunnan 650223, China 2 Key Laboratory of Computational Biology, CAS Center for Excellence in Molecular Cell Science, Collaborative Innovation Center for Genetics and Developmental Biology, Chinese Academy of Sciences-Max Planck Partner Institute for Computational Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai 200031, China 3 Yunnan Key Laboratory of Animal Reproduction, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, Yunnan 650223, China 4 University of Chinese Academy of Sciences, Beijing, China 5 Kunming College of Life Science, University of Chinese Academy of Sciences, Kunming, Yunnan 650204, China 6 Primate Research Center, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, 650223, China * Xinyi Wang and Denghui Liu contributed equally to this work 1 Downloaded from genome.cshlp.org on September 23, 2021 - Published by Cold Spring Harbor Laboratory Press # Correspondence: Jing-Dong J. Han, Email: [email protected]; Ping Zheng, Email: [email protected] Key words: rhesus monkey, pre-implantation embryo, DNA damage 2 Downloaded from genome.cshlp.org on September 23, 2021 - Published by Cold Spring Harbor Laboratory Press ABSTRACT Pre-implantation embryogenesis encompasses several critical events including genome reprogramming, zygotic genome activation (ZGA) and cell fate commitment. -

Nuclear Organization and the Epigenetic Landscape of the Mus Musculus X-Chromosome Alicia Liu University of Connecticut - Storrs, [email protected]
University of Connecticut OpenCommons@UConn Doctoral Dissertations University of Connecticut Graduate School 8-9-2019 Nuclear Organization and the Epigenetic Landscape of the Mus musculus X-Chromosome Alicia Liu University of Connecticut - Storrs, [email protected] Follow this and additional works at: https://opencommons.uconn.edu/dissertations Recommended Citation Liu, Alicia, "Nuclear Organization and the Epigenetic Landscape of the Mus musculus X-Chromosome" (2019). Doctoral Dissertations. 2273. https://opencommons.uconn.edu/dissertations/2273 Nuclear Organization and the Epigenetic Landscape of the Mus musculus X-Chromosome Alicia J. Liu, Ph.D. University of Connecticut, 2019 ABSTRACT X-linked imprinted genes have been hypothesized to contribute parent-of-origin influences on social cognition. A cluster of imprinted genes Xlr3b, Xlr4b, and Xlr4c, implicated in cognitive defects, are maternally expressed and paternally silent in the murine brain. These genes defy classic mechanisms of autosomal imprinting, suggesting a novel method of imprinted gene regulation. Using Xlr3b and Xlr4c as bait, this study uses 4C-Seq on neonatal whole brain of a 39,XO mouse model, to provide the first in-depth analysis of chromatin dynamics surrounding an imprinted locus on the X-chromosome. Significant differences in long-range contacts exist be- tween XM and XP monosomic samples. In addition, XM interaction profiles contact a greater number of genes linked to cognitive impairment, abnormality of the nervous system, and abnormality of higher mental function. This is not a pattern that is unique to the imprinted Xlr3/4 locus. Additional Alicia J. Liu - University of Connecticut - 2019 4C-Seq experiments show that other genes on the X-chromosome, implicated in intellectual disability and/or ASD, also produce more maternal contacts to other X-linked genes linked to cognitive impairment. -

Environmental Influences on Endothelial Gene Expression
ENDOTHELIAL CELL GENE EXPRESSION John Matthew Jeff Herbert Supervisors: Prof. Roy Bicknell and Dr. Victoria Heath PhD thesis University of Birmingham August 2012 University of Birmingham Research Archive e-theses repository This unpublished thesis/dissertation is copyright of the author and/or third parties. The intellectual property rights of the author or third parties in respect of this work are as defined by The Copyright Designs and Patents Act 1988 or as modified by any successor legislation. Any use made of information contained in this thesis/dissertation must be in accordance with that legislation and must be properly acknowledged. Further distribution or reproduction in any format is prohibited without the permission of the copyright holder. ABSTRACT Tumour angiogenesis is a vital process in the pathology of tumour development and metastasis. Targeting markers of tumour endothelium provide a means of targeted destruction of a tumours oxygen and nutrient supply via destruction of tumour vasculature, which in turn ultimately leads to beneficial consequences to patients. Although current anti -angiogenic and vascular targeting strategies help patients, more potently in combination with chemo therapy, there is still a need for more tumour endothelial marker discoveries as current treatments have cardiovascular and other side effects. For the first time, the analyses of in-vivo biotinylation of an embryonic system is performed to obtain putative vascular targets. Also for the first time, deep sequencing is applied to freshly isolated tumour and normal endothelial cells from lung, colon and bladder tissues for the identification of pan-vascular-targets. Integration of the proteomic, deep sequencing, public cDNA libraries and microarrays, delivers 5,892 putative vascular targets to the science community. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

140503 IPF Signatures Supplement Withfigs Thorax
Supplementary material for Heterogeneous gene expression signatures correspond to distinct lung pathologies and biomarkers of disease severity in idiopathic pulmonary fibrosis Daryle J. DePianto1*, Sanjay Chandriani1⌘*, Alexander R. Abbas1, Guiquan Jia1, Elsa N. N’Diaye1, Patrick Caplazi1, Steven E. Kauder1, Sabyasachi Biswas1, Satyajit K. Karnik1#, Connie Ha1, Zora Modrusan1, Michael A. Matthay2, Jasleen Kukreja3, Harold R. Collard2, Jackson G. Egen1, Paul J. Wolters2§, and Joseph R. Arron1§ 1Genentech Research and Early Development, South San Francisco, CA 2Department of Medicine, University of California, San Francisco, CA 3Department of Surgery, University of California, San Francisco, CA ⌘Current address: Novartis Institutes for Biomedical Research, Emeryville, CA. #Current address: Gilead Sciences, Foster City, CA. *DJD and SC contributed equally to this manuscript §PJW and JRA co-directed this project Address correspondence to Paul J. Wolters, MD University of California, San Francisco Department of Medicine Box 0111 San Francisco, CA 94143-0111 [email protected] or Joseph R. Arron, MD, PhD Genentech, Inc. MS 231C 1 DNA Way South San Francisco, CA 94080 [email protected] 1 METHODS Human lung tissue samples Tissues were obtained at UCSF from clinical samples from IPF patients at the time of biopsy or lung transplantation. All patients were seen at UCSF and the diagnosis of IPF was established through multidisciplinary review of clinical, radiological, and pathological data according to criteria established by the consensus classification of the American Thoracic Society (ATS) and European Respiratory Society (ERS), Japanese Respiratory Society (JRS), and the Latin American Thoracic Association (ALAT) (ref. 5 in main text). Non-diseased normal lung tissues were procured from lungs not used by the Northern California Transplant Donor Network. -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement. -

A Microdeletion at Xq22.2 Implicates a Glycine Receptor GLRA4 Involved in Intellectual Disability, Behavioral Problems and Craniofacial Anomalies
A microdeletion at Xq22.2 implicates a glycine receptor GLRA4 involved in intellectual disability, behavioral problems and craniofacial anomalies The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Labonne, Jonathan D. J., Tyler D. Graves, Yiping Shen, Julie R. Jones, Il-Keun Kong, Lawrence C. Layman, and Hyung-Goo Kim. 2016. “A microdeletion at Xq22.2 implicates a glycine receptor GLRA4 involved in intellectual disability, behavioral problems and craniofacial anomalies.” BMC Neurology 16 (1): 132. doi:10.1186/ s12883-016-0642-z. http://dx.doi.org/10.1186/s12883-016-0642-z. Published Version doi:10.1186/s12883-016-0642-z Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:29002418 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA Labonne et al. BMC Neurology (2016) 16:132 DOI 10.1186/s12883-016-0642-z CASE REPORT Open Access A microdeletion at Xq22.2 implicates a glycine receptor GLRA4 involved in intellectual disability, behavioral problems and craniofacial anomalies Jonathan D. J. Labonne1,2, Tyler D. Graves1, Yiping Shen3, Julie R. Jones4, Il-Keun Kong5, Lawrence C. Layman1,2,6 and Hyung-Goo Kim1,2* Abstract Background: Among the 21 annotated genes at Xq22.2, PLP1 is the only known gene involved in Xq22.2 microdeletion and microduplication syndromes with intellectual disability. Using an atypical microdeletion, which does not encompass PLP1, we implicate a novel gene GLRA4 involved in intellectual disability, behavioral problems and craniofacial anomalies. -

Gait Abnormalities and Progressive Myelin Degeneration in a New Murine Model of Pelizaeus-Merzbacher Disease with Tandem Genomic Duplication
11788 • The Journal of Neuroscience, July 17, 2013 • 33(29):11788–11799 Neurobiology of Disease Gait Abnormalities and Progressive Myelin Degeneration in a New Murine Model of Pelizaeus-Merzbacher Disease with Tandem Genomic Duplication Kristi Clark,1,2 Lauren Sakowski,1,2 Karen Sperle,1 Linda Banser,1 Carlisle P. Landel,3 Denise A. Bessert,4 Robert P. Skoff,4 and Grace M. Hobson1,2,5 1Nemours Biomedical Research, Alfred I. duPont Hospital for Children, Wilmington, Delaware 19803, 2University of Delaware, Department of Biology, Newark, Delaware 19716, 3Kimmel Cancer Center, Thomas Jefferson University, Philadelphia, Pennsylvania 19107, 4Wayne State University, Department of Anatomy and Cell Biology, Detroit, Michigan 48201, and 5Jefferson Medical College, Thomas Jefferson University, Philadelphia, Pennsylvania 19107 Pelizaeus-Merzbacher disease (PMD) is a hypomyelinating leukodystrophy caused by mutations of the proteolipid protein 1 gene (PLP1), which is located on the X chromosome and encodes the most abundant protein of myelin in the central nervous sytem. Approximately 60% of PMD cases result from genomic duplications of a region of the X chromosome that includes the entire PLP1 gene. The duplications are typically in a head-to-tail arrangement, and they vary in size and gene content. Although rodent models with extra copies of Plp1 have been developed, none contains an actual genomic rearrangement that resembles those found in PMD patients. We used mutagenic insertion chromosome engineering resources to generate the Plp1dup mouse model by introducing an X chromosome duplication in the mouse genome that contains Plp1 and five neighboring genes that are also commonly duplicated in PMD patients. The Plp1dup mice display progressive gait abnormalities compared with wild-type littermates. -

Pancreatic Progenitor Cells in Mice
PANCREATIC PROGENITOR CELLS IN MICE by Megan Hussey Cleveland A dissertation submitted to Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland July, 2014 Abstract We generated a novel transgenic mouse expressing a tdTomato fluorophore, as well as a Strep/Flag-tagged version of Ptf1a from the native Ptf1a locus. I crossed this mouse line with the well-characterized Pdx1-GFP mouse, which enabled us to visualize and sort for cells of the “tip” and “trunk” progenitor domains of the mouse pancreas. The main goal of our project was to identify previously unrecognized early transcriptional targets of Ptf1at and Pdx1. I isolated early E11.5 epithelial progenitors as well as later E13.5 tip and trunk progenitors using FACS and analyzed these populations via the GeneChip Mouse Gene 1.0 ST Array. After comparison of microarray data between tip and trunk cells, differentially- expressed genes were identified, with a focus on transcription factors, with validation by in situ hybridization. Two transcription factors, Ascl2 and Lhx1, were initially identified that had no previously known role in pancreas development, and were shown by ISH to be expressed in the expected domain of the pancreas. I performed prelimary functional studies on these two transcription factors, using lentiviral shRNAs for knock down in dorsal pancreatic bud culture. ii Acknowledgments First, I would like to thank Steven Leach for accepting me into his lab and for the 7 years of mentorship he has provided. I would like to thank Sandy Muscelli, Dave Valle, Kirby Smith, Andy McCallion and the rest of the Human Genetics department for giving me the opportunity to pursue my Ph.D. -

The Application of Mouse and Human Embryonic Stem Cells with Transcriptomics in Alternative Developmental Toxicity Tests
The application of mouse and human embryonic stem cells with transcriptomics in alternative developmental toxicity tests A bridge from model species to man Sjors Schulpen © Copyright All rights reserved. No part of this publication may be reproduced or transmitted in any form by any means without permission of the author. ISBN: 978-94-6203-861-5 Cover and layout: Maud van Deursen, Utrecht the Netherlands Production: CPI/ Wöhrmann Print Service, Zutphen the Netherlands The application of mouse and human embryonic stem cells with transcriptomics in alternative developmental toxicity tests A bridge from model species to man De toepassing van embryonale stamcellen van muis en mens met transcriptomics in alternatieve testen voor ontwikkelingstoxiciteit Een brug van modelorganisme naar de mens (met een samenvatting in het Nederlands) Proefschrift ter verkrijging van de graad van doctor aan de Universiteit Utrecht op gezag van de rector magnificus, prof.dr. G.J. van der Zwaan, ingevolge het besluit van het college voor promoties in het openbaar te verdedigen op dinsdag 7 juli 2015 des middags te 2.30 uur door Sjors Hubertus Wilhelmina Schulpen geboren op 5 januari 1983 te Sittard Promotoren: Prof. dr. A.H. Piersma Prof. dr. M. van den Berg Dit proefschrift werd mogelijk gemaakt door financiële steun van: Institute for Risk Assessment Sciences van de Universiteit Utrecht, Laboratorium voor gezondheidsbeschermingsonderzoek van het Rijksinstituut voor Volksgezondheid en Mileu, Greiner Bio-One en Stichting Proefdiervrij. Contents Abbreviations 6 -

Functionally Diverse Human T Cells Recognize Non-Microbial Antigens Presented By
RESEARCH ARTICLE Functionally diverse human T cells recognize non-microbial antigens presented by MR1 Marco Lepore1, Artem Kalinichenko1, Salvatore Calogero1, Pavanish Kumar2, Bhairav Paleja2, Mathias Schmaler1, Vipin Narang2, Francesca Zolezzi2, Michael Poidinger2,3, Lucia Mori1,2, Gennaro De Libero1,2* 1Department of Biomedicine, University Hospital and University of Basel, Basel, Switzerland; 2Singapore Immunology Network, A*STAR, Singapore, Singapore; 3Singapore Institute for Clinical Sciences, A*STAR, Singapore, Singapore Abstract MHC class I-related molecule MR1 presents riboflavin- and folate-related metabolites to mucosal-associated invariant T cells, but it is unknown whether MR1 can present alternative antigens to other T cell lineages. In healthy individuals we identified MR1-restricted T cells (named MR1T cells) displaying diverse TCRs and reacting to MR1-expressing cells in the absence of microbial ligands. Analysis of MR1T cell clones revealed specificity for distinct cell-derived antigens and alternative transcriptional strategies for metabolic programming, cell cycle control and functional polarization following antigen stimulation. Phenotypic and functional characterization of MR1T cell clones showed multiple chemokine receptor expression profiles and secretion of diverse effector molecules, suggesting functional heterogeneity. Accordingly, MR1T cells exhibited distinct T helper-like capacities upon MR1-dependent recognition of target cells expressing physiological levels of surface MR1. These data extend the role of