Functionally Enigmatic Genes: a Case Study of the Brain Ignorome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Analysis of Retinopathy in Type 1 Diabetes
Genetic Analysis of Retinopathy in Type 1 Diabetes by Sayed Mohsen Hosseini A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy Institute of Medical Science University of Toronto © Copyright by S. Mohsen Hosseini 2014 Genetic Analysis of Retinopathy in Type 1 Diabetes Sayed Mohsen Hosseini Doctor of Philosophy Institute of Medical Science University of Toronto 2014 Abstract Diabetic retinopathy (DR) is a leading cause of blindness worldwide. Several lines of evidence suggest a genetic contribution to the risk of DR; however, no genetic variant has shown convincing association with DR in genome-wide association studies (GWAS). To identify common polymorphisms associated with DR, meta-GWAS were performed in three type 1 diabetes cohorts of White subjects: Diabetes Complications and Control Trial (DCCT, n=1304), Wisconsin Epidemiologic Study of Diabetic Retinopathy (WESDR, n=603) and Renin-Angiotensin System Study (RASS, n=239). Severe (SDR) and mild (MDR) retinopathy outcomes were defined based on repeated fundus photographs in each study graded for retinopathy severity on the Early Treatment Diabetic Retinopathy Study (ETDRS) scale. Multivariable models accounted for glycemia (measured by A1C), diabetes duration and other relevant covariates in the association analyses of additive genotypes with SDR and MDR. Fixed-effects meta- analysis was used to combine the results of GWAS performed separately in WESDR, ii RASS and subgroups of DCCT, defined by cohort and treatment group. Top association signals were prioritized for replication, based on previous supporting knowledge from the literature, followed by replication in three independent white T1D studies: Genesis-GeneDiab (n=502), Steno (n=936) and FinnDiane (n=2194). -

Whole-Genome Microarray Detects Deletions and Loss of Heterozygosity of Chromosome 3 Occurring Exclusively in Metastasizing Uveal Melanoma
Anatomy and Pathology Whole-Genome Microarray Detects Deletions and Loss of Heterozygosity of Chromosome 3 Occurring Exclusively in Metastasizing Uveal Melanoma Sarah L. Lake,1 Sarah E. Coupland,1 Azzam F. G. Taktak,2 and Bertil E. Damato3 PURPOSE. To detect deletions and loss of heterozygosity of disease is fatal in 92% of patients within 2 years of diagnosis. chromosome 3 in a rare subset of fatal, disomy 3 uveal mela- Clinical and histopathologic risk factors for UM metastasis noma (UM), undetectable by fluorescence in situ hybridization include large basal tumor diameter (LBD), ciliary body involve- (FISH). ment, epithelioid cytomorphology, extracellular matrix peri- ϩ ETHODS odic acid-Schiff-positive (PAS ) loops, and high mitotic M . Multiplex ligation-dependent probe amplification 3,4 5 (MLPA) with the P027 UM assay was performed on formalin- count. Prescher et al. showed that a nonrandom genetic fixed, paraffin-embedded (FFPE) whole tumor sections from 19 change, monosomy 3, correlates strongly with metastatic death, and the correlation has since been confirmed by several disomy 3 metastasizing UMs. Whole-genome microarray analy- 3,6–10 ses using a single-nucleotide polymorphism microarray (aSNP) groups. Consequently, fluorescence in situ hybridization were performed on frozen tissue samples from four fatal dis- (FISH) detection of chromosome 3 using a centromeric probe omy 3 metastasizing UMs and three disomy 3 tumors with Ͼ5 became routine practice for UM prognostication; however, 5% years’ metastasis-free survival. to 20% of disomy 3 UM patients unexpectedly develop metas- tases.11 Attempts have therefore been made to identify the RESULTS. Two metastasizing UMs that had been classified as minimal region(s) of deletion on chromosome 3.12–15 Despite disomy 3 by FISH analysis of a small tumor sample were found these studies, little progress has been made in defining the key on MLPA analysis to show monosomy 3. -

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -
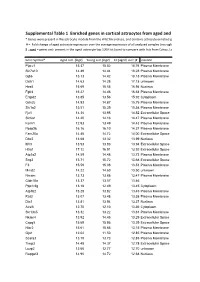
Supplemental Table 1 Enriched Genes in Cortical Astrocytes from Aged
Supplemental Table 1 Enriched genes in cortical astrocytes from aged and young-adult mice * Genes were present in the astrocyte module from the WGCNA analysis, and contains astrocyte enriched genes compared to microglia and oligodendrocytes # = Fold change of aged astrocyte expression over the average expression of all analyzed samples (microglia, astrocytes: young, old, with and without myelin contamination) $ ; aged = genes only present in the aged astrocyte top 1000 list (used to compare with lists from Cahoy, Lovatt, Doyle; see Fig. 4B), all = genes present in all astrocyte top 1000 lists Gene Symbol* Aged astr. (log2) Young astr.(log2) FC (aged/ aver.)# Location Ptprz1 15.37 15.02 18.76 Plasma Membrane Slc7a10 14.49 14.44 18.28 Plasma Membrane Gjb6 15.13 14.42 18.18 Plasma Membrane Dclk1 14.63 14.28 17.18 unknown Hes5 15.69 15.55 16.94 Nucleus Fgfr3 15.27 14.46 16.54 Plasma Membrane Entpd2 13.85 13.56 15.92 Cytoplasm Grin2c 14.93 14.87 15.75 Plasma Membrane Slc1a2 15.51 15.39 15.58 Plasma Membrane Fjx1 14.36 13.98 14.52 Extracellular Space Slc6a1 14.20 14.16 14.47 Plasma Membrane Kcnk1 12.93 13.49 14.43 Plasma Membrane Ppap2b 16.16 16.10 14.37 Plasma Membrane Fam20a 14.48 14.72 14.00 Extracellular Space Dbx2 13.68 13.32 13.99 Nucleus Itih3 13.93 13.93 13.94 Extracellular Space Htra1 17.12 16.91 13.92 Extracellular Space Atp1a2 14.59 14.48 13.73 Plasma Membrane Scg3 15.71 15.72 13.68 Extracellular Space F3 15.59 15.08 13.51 Plasma Membrane Mmd2 14.22 14.60 13.50 unknown Nrcam 13.73 13.88 13.47 Plasma Membrane Cldn10a 13.37 13.57 13.46 -

Directed Proximity-Dependent Biotin Labelling in Zebrafish
TOOLS AND RESOURCES In vivo proteomic mapping through GFP- directed proximity-dependent biotin labelling in zebrafish Zherui Xiong1, Harriet P Lo1, Kerrie-Ann McMahon1, Nick Martel1, Alun Jones1, Michelle M Hill2, Robert G Parton1,3*, Thomas E Hall1* 1Institute for Molecular Bioscience, The University of Queensland, Brisbane, Australia; 2QIMR Berghofer Medical Research Institute, Herston, Australia; 3Centre for Microscopy and Microanalysis, The University of Queensland, Brisbane, Australia Abstract Protein interaction networks are crucial for complex cellular processes. However, the elucidation of protein interactions occurring within highly specialised cells and tissues is challenging. Here, we describe the development, and application, of a new method for proximity- dependent biotin labelling in whole zebrafish. Using a conditionally stabilised GFP-binding nanobody to target a biotin ligase to GFP-labelled proteins of interest, we show tissue-specific proteomic profiling using existing GFP-tagged transgenic zebrafish lines. We demonstrate the applicability of this approach, termed BLITZ (Biotin Labelling In Tagged Zebrafish), in diverse cell types such as neurons and vascular endothelial cells. We applied this methodology to identify interactors of caveolar coat protein, cavins, in skeletal muscle. Using this system, we defined specific interaction networks within in vivo muscle cells for the closely related but functionally distinct Cavin4 and Cavin1 proteins. *For correspondence: Introduction [email protected] (RGP); [email protected] (TEH) The understanding of the biological functions of a protein requires detailed knowledge of the mole- cules with which it interacts. However, robust elucidation of interacting proteins, including not only Competing interests: The strong direct protein-protein interactions, but also weak, transient or indirect interactions is challeng- authors declare that no ing. -

New Approaches to Functional Process Discovery in HPV 16-Associated Cervical Cancer Cells by Gene Ontology
Cancer Research and Treatment 2003;35(4):304-313 New Approaches to Functional Process Discovery in HPV 16-Associated Cervical Cancer Cells by Gene Ontology Yong-Wan Kim, Ph.D.1, Min-Je Suh, M.S.1, Jin-Sik Bae, M.S.1, Su Mi Bae, M.S.1, Joo Hee Yoon, M.D.2, Soo Young Hur, M.D.2, Jae Hoon Kim, M.D.2, Duck Young Ro, M.D.2, Joon Mo Lee, M.D.2, Sung Eun Namkoong, M.D.2, Chong Kook Kim, Ph.D.3 and Woong Shick Ahn, M.D.2 1Catholic Research Institutes of Medical Science, 2Department of Obstetrics and Gynecology, College of Medicine, The Catholic University of Korea, Seoul; 3College of Pharmacy, Seoul National University, Seoul, Korea Purpose: This study utilized both mRNA differential significant genes of unknown function affected by the display and the Gene Ontology (GO) analysis to char- HPV-16-derived pathway. The GO analysis suggested that acterize the multiple interactions of a number of genes the cervical cancer cells underwent repression of the with gene expression profiles involved in the HPV-16- cancer-specific cell adhesive properties. Also, genes induced cervical carcinogenesis. belonging to DNA metabolism, such as DNA repair and Materials and Methods: mRNA differential displays, replication, were strongly down-regulated, whereas sig- with HPV-16 positive cervical cancer cell line (SiHa), and nificant increases were shown in the protein degradation normal human keratinocyte cell line (HaCaT) as a con- and synthesis. trol, were used. Each human gene has several biological Conclusion: The GO analysis can overcome the com- functions in the Gene Ontology; therefore, several func- plexity of the gene expression profile of the HPV-16- tions of each gene were chosen to establish a powerful associated pathway, identify several cancer-specific cel- cervical carcinogenesis pathway. -

Nuclear Organization and the Epigenetic Landscape of the Mus Musculus X-Chromosome Alicia Liu University of Connecticut - Storrs, [email protected]
University of Connecticut OpenCommons@UConn Doctoral Dissertations University of Connecticut Graduate School 8-9-2019 Nuclear Organization and the Epigenetic Landscape of the Mus musculus X-Chromosome Alicia Liu University of Connecticut - Storrs, [email protected] Follow this and additional works at: https://opencommons.uconn.edu/dissertations Recommended Citation Liu, Alicia, "Nuclear Organization and the Epigenetic Landscape of the Mus musculus X-Chromosome" (2019). Doctoral Dissertations. 2273. https://opencommons.uconn.edu/dissertations/2273 Nuclear Organization and the Epigenetic Landscape of the Mus musculus X-Chromosome Alicia J. Liu, Ph.D. University of Connecticut, 2019 ABSTRACT X-linked imprinted genes have been hypothesized to contribute parent-of-origin influences on social cognition. A cluster of imprinted genes Xlr3b, Xlr4b, and Xlr4c, implicated in cognitive defects, are maternally expressed and paternally silent in the murine brain. These genes defy classic mechanisms of autosomal imprinting, suggesting a novel method of imprinted gene regulation. Using Xlr3b and Xlr4c as bait, this study uses 4C-Seq on neonatal whole brain of a 39,XO mouse model, to provide the first in-depth analysis of chromatin dynamics surrounding an imprinted locus on the X-chromosome. Significant differences in long-range contacts exist be- tween XM and XP monosomic samples. In addition, XM interaction profiles contact a greater number of genes linked to cognitive impairment, abnormality of the nervous system, and abnormality of higher mental function. This is not a pattern that is unique to the imprinted Xlr3/4 locus. Additional Alicia J. Liu - University of Connecticut - 2019 4C-Seq experiments show that other genes on the X-chromosome, implicated in intellectual disability and/or ASD, also produce more maternal contacts to other X-linked genes linked to cognitive impairment. -

Environmental Influences on Endothelial Gene Expression
ENDOTHELIAL CELL GENE EXPRESSION John Matthew Jeff Herbert Supervisors: Prof. Roy Bicknell and Dr. Victoria Heath PhD thesis University of Birmingham August 2012 University of Birmingham Research Archive e-theses repository This unpublished thesis/dissertation is copyright of the author and/or third parties. The intellectual property rights of the author or third parties in respect of this work are as defined by The Copyright Designs and Patents Act 1988 or as modified by any successor legislation. Any use made of information contained in this thesis/dissertation must be in accordance with that legislation and must be properly acknowledged. Further distribution or reproduction in any format is prohibited without the permission of the copyright holder. ABSTRACT Tumour angiogenesis is a vital process in the pathology of tumour development and metastasis. Targeting markers of tumour endothelium provide a means of targeted destruction of a tumours oxygen and nutrient supply via destruction of tumour vasculature, which in turn ultimately leads to beneficial consequences to patients. Although current anti -angiogenic and vascular targeting strategies help patients, more potently in combination with chemo therapy, there is still a need for more tumour endothelial marker discoveries as current treatments have cardiovascular and other side effects. For the first time, the analyses of in-vivo biotinylation of an embryonic system is performed to obtain putative vascular targets. Also for the first time, deep sequencing is applied to freshly isolated tumour and normal endothelial cells from lung, colon and bladder tissues for the identification of pan-vascular-targets. Integration of the proteomic, deep sequencing, public cDNA libraries and microarrays, delivers 5,892 putative vascular targets to the science community. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Endoglin Protein Interactome Profiling Identifies TRIM21 and Galectin-3 As
cells Article Endoglin Protein Interactome Profiling Identifies TRIM21 and Galectin-3 as New Binding Partners 1, 1, 2, Eunate Gallardo-Vara y, Lidia Ruiz-Llorente y, Juan Casado-Vela y , 3 4 5 6, , María J. Ruiz-Rodríguez , Natalia López-Andrés , Asit K. Pattnaik , Miguel Quintanilla z * 1, , and Carmelo Bernabeu z * 1 Centro de Investigaciones Biológicas, Consejo Superior de Investigaciones Científicas (CSIC), and Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), 28040 Madrid, Spain; [email protected] (E.G.-V.); [email protected] (L.R.-L.) 2 Bioengineering and Aerospace Engineering Department, Universidad Carlos III and Centro de Investigación Biomédica en Red Enfermedades Neurodegenerativas (CIBERNED), Leganés, 28911 Madrid, Spain; [email protected] 3 Centro Nacional de Investigaciones Cardiovasculares (CNIC), 28029 Madrid, Spain; [email protected] 4 Cardiovascular Translational Research, Navarrabiomed, Complejo Hospitalario de Navarra (CHN), Universidad Pública de Navarra (UPNA), IdiSNA, 31008 Pamplona, Spain; [email protected] 5 School of Veterinary Medicine and Biomedical Sciences, and Nebraska Center for Virology, University of Nebraska-Lincoln, Lincoln, NE 68583, USA; [email protected] 6 Instituto de Investigaciones Biomédicas “Alberto Sols”, Consejo Superior de Investigaciones Científicas (CSIC), and Departamento de Bioquímica, Universidad Autónoma de Madrid (UAM), 28029 Madrid, Spain * Correspondence: [email protected] (M.Q.); [email protected] (C.B.) These authors contributed equally to this work. y Equal senior contribution. z Received: 7 August 2019; Accepted: 7 September 2019; Published: 13 September 2019 Abstract: Endoglin is a 180-kDa glycoprotein receptor primarily expressed by the vascular endothelium and involved in cardiovascular disease and cancer. -

Prediction of Meiosis-Essential Genes Based Upon the Dynamic Proteomes Responsive to Spermatogenesis
bioRxiv preprint doi: https://doi.org/10.1101/2020.02.05.936435; this version posted February 6, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. Prediction of meiosis-essential genes based upon the dynamic proteomes responsive to spermatogenesis Kailun Fang1,2,3,8, Qidan Li3,4,5,8, Yu Wei2,3,8, Jiaqi Shen6,8, Wenhui Guo3,4,5,7,8, Changyang Zhou2,3,8, Ruoxi Wu1, Wenqin Ying2, Lu Yu1,2, Jin Zi5, Yuxing Zhang3,4,5, Hui Yang2,3,9*, Siqi Liu3,4,5,9*, Charlie Degui Chen1,3,9* 1. State Key Laboratory of Molecular Biology, Shanghai Key Laboratory of Molecular Andrology, CAS Center for Excellence in Molecular Cell Science, Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, Shanghai 200031, China. 2. State Key Laboratory of Neuroscience, Key Laboratory of Primate Neurobiology, CAS Center for Excellence in Brain Science and Intelligence Technology, Shanghai Research Center for Brain Science and Brain-Inspired Intelligence, Institute of Neuroscience, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai 200031, China. 3. University of the Chinese Academy of Sciences, Beijing 100049, China. 4. CAS Key Laboratory of Genome Sciences and Information, Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing 100101, China. 5. BGI Genomics, BGI-Shenzhen, Shenzhen 518083, China. 6. United World College Changshu China, Jiangsu 215500, China. 7. Malvern College Qingdao, Shandong, 266109, China 8. -

Supplemental Information
Supplemental information Dissection of the genomic structure of the miR-183/96/182 gene. Previously, we showed that the miR-183/96/182 cluster is an intergenic miRNA cluster, located in a ~60-kb interval between the genes encoding nuclear respiratory factor-1 (Nrf1) and ubiquitin-conjugating enzyme E2H (Ube2h) on mouse chr6qA3.3 (1). To start to uncover the genomic structure of the miR- 183/96/182 gene, we first studied genomic features around miR-183/96/182 in the UCSC genome browser (http://genome.UCSC.edu/), and identified two CpG islands 3.4-6.5 kb 5’ of pre-miR-183, the most 5’ miRNA of the cluster (Fig. 1A; Fig. S1 and Seq. S1). A cDNA clone, AK044220, located at 3.2-4.6 kb 5’ to pre-miR-183, encompasses the second CpG island (Fig. 1A; Fig. S1). We hypothesized that this cDNA clone was derived from 5’ exon(s) of the primary transcript of the miR-183/96/182 gene, as CpG islands are often associated with promoters (2). Supporting this hypothesis, multiple expressed sequences detected by gene-trap clones, including clone D016D06 (3, 4), were co-localized with the cDNA clone AK044220 (Fig. 1A; Fig. S1). Clone D016D06, deposited by the German GeneTrap Consortium (GGTC) (http://tikus.gsf.de) (3, 4), was derived from insertion of a retroviral construct, rFlpROSAβgeo in 129S2 ES cells (Fig. 1A and C). The rFlpROSAβgeo construct carries a promoterless reporter gene, the β−geo cassette - an in-frame fusion of the β-galactosidase and neomycin resistance (Neor) gene (5), with a splicing acceptor (SA) immediately upstream, and a polyA signal downstream of the β−geo cassette (Fig.