Cryo-EM Structures of Human RNA Polymerase III in Its Unbound and Transcribing States
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-
![FK506-Binding Protein 12.6/1B, a Negative Regulator of [Ca2+], Rescues Memory and Restores Genomic Regulation in the Hippocampus of Aging Rats](https://docslib.b-cdn.net/cover/6136/fk506-binding-protein-12-6-1b-a-negative-regulator-of-ca2-rescues-memory-and-restores-genomic-regulation-in-the-hippocampus-of-aging-rats-16136.webp)
FK506-Binding Protein 12.6/1B, a Negative Regulator of [Ca2+], Rescues Memory and Restores Genomic Regulation in the Hippocampus of Aging Rats
This Accepted Manuscript has not been copyedited and formatted. The final version may differ from this version. A link to any extended data will be provided when the final version is posted online. Research Articles: Neurobiology of Disease FK506-Binding Protein 12.6/1b, a negative regulator of [Ca2+], rescues memory and restores genomic regulation in the hippocampus of aging rats John C. Gant1, Eric M. Blalock1, Kuey-Chu Chen1, Inga Kadish2, Olivier Thibault1, Nada M. Porter1 and Philip W. Landfield1 1Department of Pharmacology & Nutritional Sciences, University of Kentucky, Lexington, KY 40536 2Department of Cell, Developmental and Integrative Biology, University of Alabama at Birmingham, Birmingham, AL 35294 DOI: 10.1523/JNEUROSCI.2234-17.2017 Received: 7 August 2017 Revised: 10 October 2017 Accepted: 24 November 2017 Published: 18 December 2017 Author contributions: J.C.G. and P.W.L. designed research; J.C.G., E.M.B., K.-c.C., and I.K. performed research; J.C.G., E.M.B., K.-c.C., I.K., and P.W.L. analyzed data; J.C.G., E.M.B., O.T., N.M.P., and P.W.L. wrote the paper. Conflict of Interest: The authors declare no competing financial interests. NIH grants AG004542, AG033649, AG052050, AG037868 and McAlpine Foundation for Neuroscience Research Corresponding author: Philip W. Landfield, [email protected], Department of Pharmacology & Nutritional Sciences, University of Kentucky, 800 Rose Street, UKMC MS 307, Lexington, KY 40536 Cite as: J. Neurosci ; 10.1523/JNEUROSCI.2234-17.2017 Alerts: Sign up at www.jneurosci.org/cgi/alerts to receive customized email alerts when the fully formatted version of this article is published. -
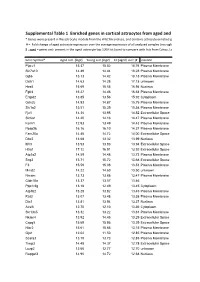
Supplemental Table 1 Enriched Genes in Cortical Astrocytes from Aged
Supplemental Table 1 Enriched genes in cortical astrocytes from aged and young-adult mice * Genes were present in the astrocyte module from the WGCNA analysis, and contains astrocyte enriched genes compared to microglia and oligodendrocytes # = Fold change of aged astrocyte expression over the average expression of all analyzed samples (microglia, astrocytes: young, old, with and without myelin contamination) $ ; aged = genes only present in the aged astrocyte top 1000 list (used to compare with lists from Cahoy, Lovatt, Doyle; see Fig. 4B), all = genes present in all astrocyte top 1000 lists Gene Symbol* Aged astr. (log2) Young astr.(log2) FC (aged/ aver.)# Location Ptprz1 15.37 15.02 18.76 Plasma Membrane Slc7a10 14.49 14.44 18.28 Plasma Membrane Gjb6 15.13 14.42 18.18 Plasma Membrane Dclk1 14.63 14.28 17.18 unknown Hes5 15.69 15.55 16.94 Nucleus Fgfr3 15.27 14.46 16.54 Plasma Membrane Entpd2 13.85 13.56 15.92 Cytoplasm Grin2c 14.93 14.87 15.75 Plasma Membrane Slc1a2 15.51 15.39 15.58 Plasma Membrane Fjx1 14.36 13.98 14.52 Extracellular Space Slc6a1 14.20 14.16 14.47 Plasma Membrane Kcnk1 12.93 13.49 14.43 Plasma Membrane Ppap2b 16.16 16.10 14.37 Plasma Membrane Fam20a 14.48 14.72 14.00 Extracellular Space Dbx2 13.68 13.32 13.99 Nucleus Itih3 13.93 13.93 13.94 Extracellular Space Htra1 17.12 16.91 13.92 Extracellular Space Atp1a2 14.59 14.48 13.73 Plasma Membrane Scg3 15.71 15.72 13.68 Extracellular Space F3 15.59 15.08 13.51 Plasma Membrane Mmd2 14.22 14.60 13.50 unknown Nrcam 13.73 13.88 13.47 Plasma Membrane Cldn10a 13.37 13.57 13.46 -

New Approaches to Functional Process Discovery in HPV 16-Associated Cervical Cancer Cells by Gene Ontology
Cancer Research and Treatment 2003;35(4):304-313 New Approaches to Functional Process Discovery in HPV 16-Associated Cervical Cancer Cells by Gene Ontology Yong-Wan Kim, Ph.D.1, Min-Je Suh, M.S.1, Jin-Sik Bae, M.S.1, Su Mi Bae, M.S.1, Joo Hee Yoon, M.D.2, Soo Young Hur, M.D.2, Jae Hoon Kim, M.D.2, Duck Young Ro, M.D.2, Joon Mo Lee, M.D.2, Sung Eun Namkoong, M.D.2, Chong Kook Kim, Ph.D.3 and Woong Shick Ahn, M.D.2 1Catholic Research Institutes of Medical Science, 2Department of Obstetrics and Gynecology, College of Medicine, The Catholic University of Korea, Seoul; 3College of Pharmacy, Seoul National University, Seoul, Korea Purpose: This study utilized both mRNA differential significant genes of unknown function affected by the display and the Gene Ontology (GO) analysis to char- HPV-16-derived pathway. The GO analysis suggested that acterize the multiple interactions of a number of genes the cervical cancer cells underwent repression of the with gene expression profiles involved in the HPV-16- cancer-specific cell adhesive properties. Also, genes induced cervical carcinogenesis. belonging to DNA metabolism, such as DNA repair and Materials and Methods: mRNA differential displays, replication, were strongly down-regulated, whereas sig- with HPV-16 positive cervical cancer cell line (SiHa), and nificant increases were shown in the protein degradation normal human keratinocyte cell line (HaCaT) as a con- and synthesis. trol, were used. Each human gene has several biological Conclusion: The GO analysis can overcome the com- functions in the Gene Ontology; therefore, several func- plexity of the gene expression profile of the HPV-16- tions of each gene were chosen to establish a powerful associated pathway, identify several cancer-specific cel- cervical carcinogenesis pathway. -

Genetic Basis of Simple and Complex Traits with Relevance to Avian Evolution
Genetic basis of simple and complex traits with relevance to avian evolution Małgorzata Anna Gazda Doctoral Program in Biodiversity, Genetics and Evolution D Faculdade de Ciências da Universidade do Porto 2019 Supervisor Miguel Jorge Pinto Carneiro, Auxiliary Researcher, CIBIO/InBIO, Laboratório Associado, Universidade do Porto Co-supervisor Ricardo Lopes, CIBIO/InBIO Leif Andersson, Uppsala University FCUP Genetic basis of avian traits Nota Previa Na elaboração desta tese, e nos termos do número 2 do Artigo 4º do Regulamento Geral dos Terceiros Ciclos de Estudos da Universidade do Porto e do Artigo 31º do D.L.74/2006, de 24 de Março, com a nova redação introduzida pelo D.L. 230/2009, de 14 de Setembro, foi efetuado o aproveitamento total de um conjunto coerente de trabalhos de investigação já publicados ou submetidos para publicação em revistas internacionais indexadas e com arbitragem científica, os quais integram alguns dos capítulos da presente tese. Tendo em conta que os referidos trabalhos foram realizados com a colaboração de outros autores, o candidato esclarece que, em todos eles, participou ativamente na sua conceção, na obtenção, análise e discussão de resultados, bem como na elaboração da sua forma publicada. Este trabalho foi apoiado pela Fundação para a Ciência e Tecnologia (FCT) através da atribuição de uma bolsa de doutoramento (PD/BD/114042/2015) no âmbito do programa doutoral em Biodiversidade, Genética e Evolução (BIODIV). 2 FCUP Genetic basis of avian traits Acknowledgements Firstly, I would like to thank to my all supervisors Miguel Carneiro, Ricardo Lopes and Leif Andersson, for the demanding task of supervising myself last four years. -

Proteomics Provides Insights Into the Inhibition of Chinese Hamster V79
www.nature.com/scientificreports OPEN Proteomics provides insights into the inhibition of Chinese hamster V79 cell proliferation in the deep underground environment Jifeng Liu1,2, Tengfei Ma1,2, Mingzhong Gao3, Yilin Liu4, Jun Liu1, Shichao Wang2, Yike Xie2, Ling Wang2, Juan Cheng2, Shixi Liu1*, Jian Zou1,2*, Jiang Wu2, Weimin Li2 & Heping Xie2,3,5 As resources in the shallow depths of the earth exhausted, people will spend extended periods of time in the deep underground space. However, little is known about the deep underground environment afecting the health of organisms. Hence, we established both deep underground laboratory (DUGL) and above ground laboratory (AGL) to investigate the efect of environmental factors on organisms. Six environmental parameters were monitored in the DUGL and AGL. Growth curves were recorded and tandem mass tag (TMT) proteomics analysis were performed to explore the proliferative ability and diferentially abundant proteins (DAPs) in V79 cells (a cell line widely used in biological study in DUGLs) cultured in the DUGL and AGL. Parallel Reaction Monitoring was conducted to verify the TMT results. γ ray dose rate showed the most detectable diference between the two laboratories, whereby γ ray dose rate was signifcantly lower in the DUGL compared to the AGL. V79 cell proliferation was slower in the DUGL. Quantitative proteomics detected 980 DAPs (absolute fold change ≥ 1.2, p < 0.05) between V79 cells cultured in the DUGL and AGL. Of these, 576 proteins were up-regulated and 404 proteins were down-regulated in V79 cells cultured in the DUGL. KEGG pathway analysis revealed that seven pathways (e.g. -

Nuclear Factor I/B Is an Oncogene in Small Cell Lung Cancer
Nuclear Factor I/B is an Oncogene in Small Cell Lung Cancer The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Dooley, A. L. et al. “Nuclear factor I/B is an oncogene in small cell lung cancer.” Genes & Development 25 (2011): 1470-1475. As Published http://dx.doi.org/10.1101/gad.2046711 Publisher Cold Spring Harbor Laboratory Press in association with The Genetics Society Version Author's final manuscript Citable link http://hdl.handle.net/1721.1/66512 Terms of Use Creative Commons Attribution-Noncommercial-Share Alike 3.0 Detailed Terms http://creativecommons.org/licenses/by-nc-sa/3.0/ Dooley 1 Nuclear Factor I/B is an Oncogene in Small Cell Lung Cancer Alison L. Dooley1, Monte M. Winslow1, Derek Y. Chiang2,3,4, Shantanu Banerji2,3, Nicolas Stransky2, Talya L. Dayton1, Eric L. Snyder1, Stephanie Senna1, Charles A. Whittaker1, Roderick T. Bronson5, Denise Crowley1, Jordi Barretina2,3, Levi Garraway2,3, Matthew Meyerson2,3, Tyler Jacks1,6 1David H. Koch Institute for Integrative Cancer Research and Department of Biology, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA 2The Broad Institute, Cancer Program, Cambridge, Massachusetts, USA 3Dana-Farber Cancer Institute, Department of Medical Oncology and Center for Cancer Genome Discovery, Boston, Massachusetts, USA 4Current address: Lineberger Comprehensive Cancer Center, 450 West Drive, CB #7295, Chapel Hill, North Carolina, USA 5Department of Pathology, Tufts University School of Medicine and Veterinary Medicine, North Grafton, Massachusetts, USA 6Howard Hughes Medical Institute, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA Key Words: Small Cell Lung Cancer, Mouse model, Nuclear Factor I/B Dooley 2 Abstract Small cell lung cancer (SCLC) is an aggressive cancer often diagnosed after it has metastasized. -

Supplemental Table 1. Complete Gene Lists and GO Terms from Figure 3C
Supplemental Table 1. Complete gene lists and GO terms from Figure 3C. Path 1 Genes: RP11-34P13.15, RP4-758J18.10, VWA1, CHD5, AZIN2, FOXO6, RP11-403I13.8, ARHGAP30, RGS4, LRRN2, RASSF5, SERTAD4, GJC2, RHOU, REEP1, FOXI3, SH3RF3, COL4A4, ZDHHC23, FGFR3, PPP2R2C, CTD-2031P19.4, RNF182, GRM4, PRR15, DGKI, CHMP4C, CALB1, SPAG1, KLF4, ENG, RET, GDF10, ADAMTS14, SPOCK2, MBL1P, ADAM8, LRP4-AS1, CARNS1, DGAT2, CRYAB, AP000783.1, OPCML, PLEKHG6, GDF3, EMP1, RASSF9, FAM101A, STON2, GREM1, ACTC1, CORO2B, FURIN, WFIKKN1, BAIAP3, TMC5, HS3ST4, ZFHX3, NLRP1, RASD1, CACNG4, EMILIN2, L3MBTL4, KLHL14, HMSD, RP11-849I19.1, SALL3, GADD45B, KANK3, CTC- 526N19.1, ZNF888, MMP9, BMP7, PIK3IP1, MCHR1, SYTL5, CAMK2N1, PINK1, ID3, PTPRU, MANEAL, MCOLN3, LRRC8C, NTNG1, KCNC4, RP11, 430C7.5, C1orf95, ID2-AS1, ID2, GDF7, KCNG3, RGPD8, PSD4, CCDC74B, BMPR2, KAT2B, LINC00693, ZNF654, FILIP1L, SH3TC1, CPEB2, NPFFR2, TRPC3, RP11-752L20.3, FAM198B, TLL1, CDH9, PDZD2, CHSY3, GALNT10, FOXQ1, ATXN1, ID4, COL11A2, CNR1, GTF2IP4, FZD1, PAX5, RP11-35N6.1, UNC5B, NKX1-2, FAM196A, EBF3, PRRG4, LRP4, SYT7, PLBD1, GRASP, ALX1, HIP1R, LPAR6, SLITRK6, C16orf89, RP11-491F9.1, MMP2, B3GNT9, NXPH3, TNRC6C-AS1, LDLRAD4, NOL4, SMAD7, HCN2, PDE4A, KANK2, SAMD1, EXOC3L2, IL11, EMILIN3, KCNB1, DOK5, EEF1A2, A4GALT, ADGRG2, ELF4, ABCD1 Term Count % PValue Genes regulation of pathway-restricted GDF3, SMAD7, GDF7, BMPR2, GDF10, GREM1, BMP7, LDLRAD4, SMAD protein phosphorylation 9 6.34 1.31E-08 ENG pathway-restricted SMAD protein GDF3, SMAD7, GDF7, BMPR2, GDF10, GREM1, BMP7, LDLRAD4, phosphorylation -

Whole Exome Sequencing in Families at High Risk for Hodgkin Lymphoma: Identification of a Predisposing Mutation in the KDR Gene
Hodgkin Lymphoma SUPPLEMENTARY APPENDIX Whole exome sequencing in families at high risk for Hodgkin lymphoma: identification of a predisposing mutation in the KDR gene Melissa Rotunno, 1 Mary L. McMaster, 1 Joseph Boland, 2 Sara Bass, 2 Xijun Zhang, 2 Laurie Burdett, 2 Belynda Hicks, 2 Sarangan Ravichandran, 3 Brian T. Luke, 3 Meredith Yeager, 2 Laura Fontaine, 4 Paula L. Hyland, 1 Alisa M. Goldstein, 1 NCI DCEG Cancer Sequencing Working Group, NCI DCEG Cancer Genomics Research Laboratory, Stephen J. Chanock, 5 Neil E. Caporaso, 1 Margaret A. Tucker, 6 and Lynn R. Goldin 1 1Genetic Epidemiology Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 2Cancer Genomics Research Laboratory, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 3Ad - vanced Biomedical Computing Center, Leidos Biomedical Research Inc.; Frederick National Laboratory for Cancer Research, Frederick, MD; 4Westat, Inc., Rockville MD; 5Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; and 6Human Genetics Program, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD, USA ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2015.135475 Received: August 19, 2015. Accepted: January 7, 2016. Pre-published: June 13, 2016. Correspondence: [email protected] Supplemental Author Information: NCI DCEG Cancer Sequencing Working Group: Mark H. Greene, Allan Hildesheim, Nan Hu, Maria Theresa Landi, Jennifer Loud, Phuong Mai, Lisa Mirabello, Lindsay Morton, Dilys Parry, Anand Pathak, Douglas R. Stewart, Philip R. Taylor, Geoffrey S. Tobias, Xiaohong R. Yang, Guoqin Yu NCI DCEG Cancer Genomics Research Laboratory: Salma Chowdhury, Michael Cullen, Casey Dagnall, Herbert Higson, Amy A. -

Supplementary Data
Supplementary Fig. 1 A B Responder_Xenograft_ Responder_Xenograft_ NON- NON- Lu7336, Vehicle vs Lu7466, Vehicle vs Responder_Xenograft_ Responder_Xenograft_ Sagopilone, Welch- Sagopilone, Welch- Lu7187, Vehicle vs Lu7406, Vehicle vs Test: 638 Test: 600 Sagopilone, Welch- Sagopilone, Welch- Test: 468 Test: 482 Responder_Xenograft_ NON- Lu7860, Vehicle vs Responder_Xenograft_ Sagopilone, Welch - Lu7558, Vehicle vs Test: 605 Sagopilone, Welch- Test: 333 Supplementary Fig. 2 Supplementary Fig. 3 Supplementary Figure S1. Venn diagrams comparing probe sets regulated by Sagopilone treatment (10mg/kg for 24h) between individual models (Welsh Test ellipse p-value<0.001 or 5-fold change). A Sagopilone responder models, B Sagopilone non-responder models. Supplementary Figure S2. Pathway analysis of genes regulated by Sagopilone treatment in responder xenograft models 24h after Sagopilone treatment by GeneGo Metacore; the most significant pathway map representing cell cycle/spindle assembly and chromosome separation is shown, genes upregulated by Sagopilone treatment are marked with red thermometers. Supplementary Figure S3. GeneGo Metacore pathway analysis of genes differentially expressed between Sagopilone Responder and Non-Responder models displaying –log(p-Values) of most significant pathway maps. Supplementary Tables Supplementary Table 1. Response and activity in 22 non-small-cell lung cancer (NSCLC) xenograft models after treatment with Sagopilone and other cytotoxic agents commonly used in the management of NSCLC Tumor Model Response type -

Original Article a Database and Functional Annotation of NF-Κb Target Genes
Int J Clin Exp Med 2016;9(5):7986-7995 www.ijcem.com /ISSN:1940-5901/IJCEM0019172 Original Article A database and functional annotation of NF-κB target genes Yang Yang, Jian Wu, Jinke Wang The State Key Laboratory of Bioelectronics, Southeast University, Nanjing 210096, People’s Republic of China Received November 4, 2015; Accepted February 10, 2016; Epub May 15, 2016; Published May 30, 2016 Abstract: Backgrounds: The previous studies show that the transcription factor NF-κB always be induced by many inducers, and can regulate the expressions of many genes. The aim of the present study is to explore the database and functional annotation of NF-κB target genes. Methods: In this study, we manually collected the most complete listing of all NF-κB target genes identified to date, including the NF-κB microRNA target genes and built the database of NF-κB target genes with the detailed information of each target gene and annotated it by DAVID tools. Results: The NF-κB target genes database was established (http://tfdb.seu.edu.cn/nfkb/). The collected data confirmed that NF-κB maintains multitudinous biological functions and possesses the considerable complexity and diversity in regulation the expression of corresponding target genes set. The data showed that the NF-κB was a central regula- tor of the stress response, immune response and cellular metabolic processes. NF-κB involved in bone disease, immunological disease and cardiovascular disease, various cancers and nervous disease. NF-κB can modulate the expression activity of other transcriptional factors. Inhibition of IKK and IκBα phosphorylation, the decrease of nuclear translocation of p65 and the reduction of intracellular glutathione level determined the up-regulation or down-regulation of expression of NF-κB target genes. -

Genomic Study of RNA Polymerase II and III Snapc-Bound Promoters Reveals a Gene Transcribed by Both Enzymes and a Broad Use of Common Activators
Genomic Study of RNA Polymerase II and III SNAPc-Bound Promoters Reveals a Gene Transcribed by Both Enzymes and a Broad Use of Common Activators Nicole James Faresse1., Donatella Canella1., Viviane Praz1,2, Joe¨lle Michaud1¤, David Romascano1, Nouria Hernandez1* 1 Center for Integrative Genomics, Faculty of Biology and Medicine, University of Lausanne, Lausanne, Switzerland, 2 Swiss Institute of Bioinformatics, Lausanne, Switzerland Abstract SNAPc is one of a few basal transcription factors used by both RNA polymerase (pol) II and pol III. To define the set of active SNAPc-dependent promoters in human cells, we have localized genome-wide four SNAPc subunits, GTF2B (TFIIB), BRF2, pol II, and pol III. Among some seventy loci occupied by SNAPc and other factors, including pol II snRNA genes, pol III genes with type 3 promoters, and a few un-annotated loci, most are primarily occupied by either pol II and GTF2B, or pol III and BRF2. A notable exception is the RPPH1 gene, which is occupied by significant amounts of both polymerases. We show that the large majority of SNAPc-dependent promoters recruit POU2F1 and/or ZNF143 on their enhancer region, and a subset also recruits GABP, a factor newly implicated in SNAPc-dependent transcription. These activators associate with pol II and III promoters in G1 slightly before the polymerase, and ZNF143 is required for efficient transcription initiation complex assembly. The results characterize a set of genes with unique properties and establish that polymerase specificity is not absolute in vivo. Citation: James Faresse N, Canella D, Praz V, Michaud J, Romascano D, et al. -

Genomic Anatomy of the Tyrp1 (Brown) Deletion Complex
Genomic anatomy of the Tyrp1 (brown) deletion complex Ian M. Smyth*, Laurens Wilming†, Angela W. Lee*, Martin S. Taylor*, Phillipe Gautier*, Karen Barlow†, Justine Wallis†, Sancha Martin†, Rebecca Glithero†, Ben Phillimore†, Sarah Pelan†, Rob Andrew†, Karen Holt†, Ruth Taylor†, Stuart McLaren†, John Burton†, Jonathon Bailey†, Sarah Sims†, Jan Squares†, Bob Plumb†, Ann Joy†, Richard Gibson†, James Gilbert†, Elizabeth Hart†, Gavin Laird†, Jane Loveland†, Jonathan Mudge†, Charlie Steward†, David Swarbreck†, Jennifer Harrow†, Philip North‡, Nicholas Leaves‡, John Greystrong‡, Maria Coppola‡, Shilpa Manjunath‡, Mark Campbell‡, Mark Smith‡, Gregory Strachan‡, Calli Tofts‡, Esther Boal‡, Victoria Cobley‡, Giselle Hunter‡, Christopher Kimberley‡, Daniel Thomas‡, Lee Cave-Berry‡, Paul Weston‡, Marc R. M. Botcherby‡, Sharon White*, Ruth Edgar*, Sally H. Cross*, Marjan Irvani¶, Holger Hummerich¶, Eleanor H. Simpson*, Dabney Johnson§, Patricia R. Hunsicker§, Peter F. R. Little¶, Tim Hubbard†, R. Duncan Campbell‡, Jane Rogers†, and Ian J. Jackson*ʈ *Medical Research Council Human Genetics Unit, Edinburgh EH4 2XU, United Kingdom; †Wellcome Trust Sanger Institute, and ‡Medical Research Council Rosalind Franklin Centre for Genome Research, Hinxton CB10 1SA, United Kingdom; §Life Sciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831; and ¶Department of Biochemistry, Imperial College, London SW7 2AZ, United Kingdom Communicated by Liane B. Russell, Oak Ridge National Laboratory, Oak Ridge, TN, January 9, 2006 (received for review September 15, 2005) Chromosome deletions in the mouse have proven invaluable in the deletions also provided the means to produce physical maps of dissection of gene function. The brown deletion complex com- genetic markers. Studies of this kind have been published for prises >28 independent genome rearrangements, which have several loci, including albino (Tyr), piebald (Ednrb), pink-eyed been used to identify several functional loci on chromosome 4 dilution (p), and the brown deletion complex (2–6).