Egfr Activates a Taz-Driven Oncogenic Program in Glioblastoma
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Association Analyses of Known Genetic Variants with Gene
ASSOCIATION ANALYSES OF KNOWN GENETIC VARIANTS WITH GENE EXPRESSION IN BRAIN by Viktoriya Strumba A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Bioinformatics) in The University of Michigan 2009 Doctoral Committee: Professor Margit Burmeister, Chair Professor Huda Akil Professor Brian D. Athey Assistant Professor Zhaohui S. Qin Research Statistician Thomas Blackwell To Sam and Valentina Dmitriy and Elizabeth ii ACKNOWLEDGEMENTS I would like to thank my advisor Professor Margit Burmeister, who tirelessly guided me though seemingly impassable corridors of graduate work. Throughout my thesis writing period she provided sound advice, encouragement and inspiration. Leading by example, her enthusiasm and dedication have been instrumental in my path to becoming a better scientist. I also would like to thank my co-advisor Tom Blackwell. His careful prodding always kept me on my toes and looking for answers, which taught me the depth of careful statistical analysis. His diligence and dedication have been irreplaceable in most difficult of projects. I also would like to thank my other committee members: Huda Akil, Brian Athey and Steve Qin as well as David States. You did not make it easy for me, but I thank you for believing and not giving up. Huda’s eloquence in every subject matter she explained have been particularly inspiring, while both Huda’s and Brian’s valuable advice made the completion of this dissertation possible. I would also like to thank all the members of the Burmeister lab, both past and present: Sandra Villafuerte, Kristine Ito, Cindy Schoen, Karen Majczenko, Ellen Schmidt, Randi Burns, Gang Su, Nan Xiang and Ana Progovac. -

Structure and Function of the Fgd Family of Divergent FYVE Domain Proteins
Biochemistry and Cell Biology Structure and Function of the Fgd Family of Divergent FYVE Domain Proteins Journal: Biochemistry and Cell Biology Manuscript ID bcb-2018-0185.R1 Manuscript Type: Mini Review Date Submitted by the 03-Aug-2018 Author: Complete List of Authors: Eitzen, Gary; University of Alberta Faculty of Medicine and Dentistry Smithers, Cameron C.; University of Alberta, Biochemistry Murray, Allan; University of Alberta Faculty of Medicine and Dentistry Overduin, Michael; University of Alberta Faculty of Medicine and Dentistry Draft Fgd, Pleckstrin Homology domain, FYVE domain, Dbl Homology Domain, Keyword: Rho GEF Is the invited manuscript for consideration in a Special CSMB Special Issue Issue? : https://mc06.manuscriptcentral.com/bcb-pubs Page 1 of 37 Biochemistry and Cell Biology Title: Structure and Function of the Fgd Family of Divergent FYVE Domain Proteins Authors: Gary Eitzen1, Cameron C. Smithers2, Allan G Murray3 and Michael Overduin2* Draft 1Department of Cell Biology, 2Department of Biochemistry, 3Department of Medicine, University of Alberta, Edmonton, Alberta, Canada *Corresponding author. Michael Overduin Telephone: +1 780 492 3518 Fax: +1 780 492-0886 E-mail: [email protected] https://mc06.manuscriptcentral.com/bcb-pubs Biochemistry and Cell Biology Page 2 of 37 Abstract FYVE domains are highly conserved protein modules that typically bind phosphatidylinositol 3-phosphate (PI3P) on the surface of early endosomes. Along with pleckstrin homology (PH) and phox homology (PX) domains, FYVE domains are the principal readers of the phosphoinositide (PI) code that mediate specific recognition of eukaryotic organelles. Of all the human FYVE domain-containing proteins, those within the Faciogenital dysplasia (Fgd) subfamily are particularly divergent, and couple with GTPases to exert unique cellular functions. -
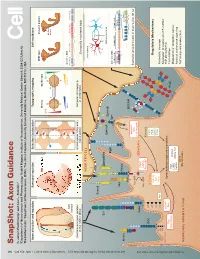
Snapshot: Axon Guidance Pasterkamp R
494 1 Cell Cell ??? SnapShot: Axon Guidance 153 SnapShot: XXXXXXXXXXXXXXXXXXXXXXXXXX 1 2 , ??MONTH?? ??DATE??, 200? ©200? Elsevier Inc. 200?©200? ElsevierInc. , ??MONTH?? ??DATE??, DOI R. Jeroen Pasterkamp and Alex L. Kolodkin , April11, 2013©2013Elsevier Inc. DOI http://dx.doi.org/10.1016/j.cell.2013.03.031 AUTHOR XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX 1 AFFILIATIONDepartment of XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX Neuroscience and Pharmacology, Rudolf Magnus Institute of Neuroscience, University Medical Center Utrecht, 3584 CG Utrecht, The Netherlands; 2Department of Neuroscience, HHMI, The Johns Hopkins University School of Medicine, Baltimore, MD 21212, USA Axon attraction and repulsion Surround repulsion Selective fasciculation Topographic mapping Self-avoidance Wild-type Dscam1 mutant Sema3A A Retina SC/Tectum EphB EphrinB P D D Mutant T A neuron N P A V V Genomic DNA P EphA EphrinA Slit Exon 4 (12) Exon 6 (48) Exon 9 (33) Exon 17 (2) Netrin Commissural axon guidance Surround repulsion of peripheral Grasshopper CNS axon Retinotectal mapping at the CNS midline nerves in vertebrates fasciculation in vertebrates Drosophila mushroom body XXXXXXXXX NEURITE/CELL Isoneuronal Sema3 Slit Sema1/4-6 Heteroneuronal EphrinA EphrinB FasII Eph Genomic DNA Pcdh-α (14) Pcdh-β (22) Pcdh-γ (22) Variable Con Variable Con Nrp Plexin ** *** Netrin LAMELLIPODIA Con DSCAM ephexin Starburst amacrine cells in mammalian retina Ras-GTP Vav See online version for legend and references. α-chimaerin GEFs/GAPs Robo FARP Ras-GDP LARG RhoGEF Kinases DCC cc0 GTPases PKA cc1 FAK Regulatory Mechanisms See online versionfor??????. Cdc42 GSK3 cc2 Rac PI3K P1 Rho P2 cc3 Abl Proteolytic cleavage P3 Regulation of expression (TF, miRNA, FILOPODIA srGAP Cytoskeleton regulatory proteins multiple isoforms) Sos Trio Pcdh Cis inhibition DOCK180 PAK ROCK Modulation of receptors’ output LIMK Myosin-II Colin Forward and reverse signaling Actin Trafcking and endocytosis NEURONAL GROWTH CONE Microtubules SnapShot: Axon Guidance R. -

Regulation of Cdc42 and Its Effectors in Epithelial Morphogenesis Franck Pichaud1,2,*, Rhian F
© 2019. Published by The Company of Biologists Ltd | Journal of Cell Science (2019) 132, jcs217869. doi:10.1242/jcs.217869 REVIEW SUBJECT COLLECTION: ADHESION Regulation of Cdc42 and its effectors in epithelial morphogenesis Franck Pichaud1,2,*, Rhian F. Walther1 and Francisca Nunes de Almeida1 ABSTRACT An overview of Cdc42 Cdc42 – a member of the small Rho GTPase family – regulates cell Cdc42 was discovered in yeast and belongs to a large family of small – polarity across organisms from yeast to humans. It is an essential (20 30 kDa) GTP-binding proteins (Adams et al., 1990; Johnson regulator of polarized morphogenesis in epithelial cells, through and Pringle, 1990). It is part of the Ras-homologous Rho subfamily coordination of apical membrane morphogenesis, lumen formation and of GTPases, of which there are 20 members in humans, including junction maturation. In parallel, work in yeast and Caenorhabditis elegans the RhoA and Rac GTPases, (Hall, 2012). Rho, Rac and Cdc42 has provided important clues as to how this molecular switch can homologues are found in all eukaryotes, except for plants, which do generate and regulate polarity through localized activation or inhibition, not have a clear homologue for Cdc42. Together, the function of and cytoskeleton regulation. Recent studies have revealed how Rho GTPases influences most, if not all, cellular processes. important and complex these regulations can be during epithelial In the early 1990s, seminal work from Alan Hall and his morphogenesis. This complexity is mirrored by the fact that Cdc42 can collaborators identified Rho, Rac and Cdc42 as main regulators of exert its function through many effector proteins. -

Efficacy and Mechanistic Evaluation of Tic10, a Novel Antitumor Agent
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2012 Efficacy and Mechanisticv E aluation of Tic10, A Novel Antitumor Agent Joshua Edward Allen University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Oncology Commons Recommended Citation Allen, Joshua Edward, "Efficacy and Mechanisticv E aluation of Tic10, A Novel Antitumor Agent" (2012). Publicly Accessible Penn Dissertations. 488. https://repository.upenn.edu/edissertations/488 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/488 For more information, please contact [email protected]. Efficacy and Mechanisticv E aluation of Tic10, A Novel Antitumor Agent Abstract TNF-related apoptosis-inducing ligand (TRAIL; Apo2L) is an endogenous protein that selectively induces apoptosis in cancer cells and is a critical effector in the immune surveillance of cancer. Recombinant TRAIL and TRAIL-agonist antibodies are in clinical trials for the treatment of solid malignancies due to the cancer-specific cytotoxicity of TRAIL. Recombinant TRAIL has a short serum half-life and both recombinant TRAIL and TRAIL receptor agonist antibodies have a limited capacity to perfuse to tissue compartments such as the brain, limiting their efficacy in certain malignancies. To overcome such limitations, we searched for small molecules capable of inducing the TRAIL gene using a high throughput luciferase reporter gene assay. We selected TRAIL-inducing compound 10 (TIC10) for further study based on its induction of TRAIL at the cell surface and its promising therapeutic index. TIC10 is a potent, stable, and orally active antitumor agent that crosses the blood-brain barrier and transcriptionally induces TRAIL and TRAIL-mediated cell death in a p53-independent manner. -

Regulation of the Mammalian Target of Rapamycin Complex 2 (Mtorc2)
Regulation of the Mammalian Target Of Rapamycin Complex 2 (mTORC2) Inauguraldissertation Zur Erlangung der Würde eines Doktors der Philosophie vorgelegt der Philosophisch-Naturwissenschaftlichen Fakultät der Universität Basel von Klaus-Dieter Molle aus Heilbronn, Deutschland Basel, 2006 Genehmigt von der Philosophisch-Naturwissenschaftlichen Fakultät Auf Antrag von Prof. Michael N. Hall und Prof. Markus Affolter. Basel, den 21.11.2006 Prof. Hans-Peter Hauri Dekan Summary The growth controlling mammalian Target of Rapamycin (mTOR) is a conserved Ser/Thr kinase found in two structurally and functionally distinct complexes, mTORC1 and mTORC2. The tumor suppressor TSC1-TSC2 complex inhibits mTORC1 by acting on the small GTPase Rheb, but the role of TSC1-TSC2 and Rheb in the regulation of mTORC2 is unclear. Here we examined the role of TSC1-TSC2 in the regulation of mTORC2 in human embryonic kidney 293 cells. Induced knockdown of TSC1 and TSC2 (TSC1/2) stimulated mTORC2-dependent actin cytoskeleton organization and Paxillin phosphorylation. Furthermore, TSC1/2 siRNA increased mTORC2-dependent Ser473 phosphorylation of plasma membrane bound, myristoylated Akt/PKB. This suggests that loss of Akt/PKB Ser473 phosphorylation in TSC mutant cells, as reported previously, is due to inhibition of Akt/PKB localization rather than inhibition of mTORC2 activity. Amino acids and overexpression of Rheb failed to stimulate mTORC2 signaling. Thus, TSC1-TSC2 also inhibits mTORC2, but possibly independently of Rheb. Our results suggest that mTORC2 hyperactivation may contribute to the pathophysiology of diseases such as cancer and Tuberous Sclerosis Complex. i Acknowledgement During my PhD studies in the Biozentrum I received a lot of support from many people around me who I mention here to express my gratefulness. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Investigation of Candidate Genes and Mechanisms Underlying Obesity
Prashanth et al. BMC Endocrine Disorders (2021) 21:80 https://doi.org/10.1186/s12902-021-00718-5 RESEARCH ARTICLE Open Access Investigation of candidate genes and mechanisms underlying obesity associated type 2 diabetes mellitus using bioinformatics analysis and screening of small drug molecules G. Prashanth1 , Basavaraj Vastrad2 , Anandkumar Tengli3 , Chanabasayya Vastrad4* and Iranna Kotturshetti5 Abstract Background: Obesity associated type 2 diabetes mellitus is a metabolic disorder ; however, the etiology of obesity associated type 2 diabetes mellitus remains largely unknown. There is an urgent need to further broaden the understanding of the molecular mechanism associated in obesity associated type 2 diabetes mellitus. Methods: To screen the differentially expressed genes (DEGs) that might play essential roles in obesity associated type 2 diabetes mellitus, the publicly available expression profiling by high throughput sequencing data (GSE143319) was downloaded and screened for DEGs. Then, Gene Ontology (GO) and REACTOME pathway enrichment analysis were performed. The protein - protein interaction network, miRNA - target genes regulatory network and TF-target gene regulatory network were constructed and analyzed for identification of hub and target genes. The hub genes were validated by receiver operating characteristic (ROC) curve analysis and RT- PCR analysis. Finally, a molecular docking study was performed on over expressed proteins to predict the target small drug molecules. Results: A total of 820 DEGs were identified between -

Supplemental Table 1. Complete Gene Lists and GO Terms from Figure 3C
Supplemental Table 1. Complete gene lists and GO terms from Figure 3C. Path 1 Genes: RP11-34P13.15, RP4-758J18.10, VWA1, CHD5, AZIN2, FOXO6, RP11-403I13.8, ARHGAP30, RGS4, LRRN2, RASSF5, SERTAD4, GJC2, RHOU, REEP1, FOXI3, SH3RF3, COL4A4, ZDHHC23, FGFR3, PPP2R2C, CTD-2031P19.4, RNF182, GRM4, PRR15, DGKI, CHMP4C, CALB1, SPAG1, KLF4, ENG, RET, GDF10, ADAMTS14, SPOCK2, MBL1P, ADAM8, LRP4-AS1, CARNS1, DGAT2, CRYAB, AP000783.1, OPCML, PLEKHG6, GDF3, EMP1, RASSF9, FAM101A, STON2, GREM1, ACTC1, CORO2B, FURIN, WFIKKN1, BAIAP3, TMC5, HS3ST4, ZFHX3, NLRP1, RASD1, CACNG4, EMILIN2, L3MBTL4, KLHL14, HMSD, RP11-849I19.1, SALL3, GADD45B, KANK3, CTC- 526N19.1, ZNF888, MMP9, BMP7, PIK3IP1, MCHR1, SYTL5, CAMK2N1, PINK1, ID3, PTPRU, MANEAL, MCOLN3, LRRC8C, NTNG1, KCNC4, RP11, 430C7.5, C1orf95, ID2-AS1, ID2, GDF7, KCNG3, RGPD8, PSD4, CCDC74B, BMPR2, KAT2B, LINC00693, ZNF654, FILIP1L, SH3TC1, CPEB2, NPFFR2, TRPC3, RP11-752L20.3, FAM198B, TLL1, CDH9, PDZD2, CHSY3, GALNT10, FOXQ1, ATXN1, ID4, COL11A2, CNR1, GTF2IP4, FZD1, PAX5, RP11-35N6.1, UNC5B, NKX1-2, FAM196A, EBF3, PRRG4, LRP4, SYT7, PLBD1, GRASP, ALX1, HIP1R, LPAR6, SLITRK6, C16orf89, RP11-491F9.1, MMP2, B3GNT9, NXPH3, TNRC6C-AS1, LDLRAD4, NOL4, SMAD7, HCN2, PDE4A, KANK2, SAMD1, EXOC3L2, IL11, EMILIN3, KCNB1, DOK5, EEF1A2, A4GALT, ADGRG2, ELF4, ABCD1 Term Count % PValue Genes regulation of pathway-restricted GDF3, SMAD7, GDF7, BMPR2, GDF10, GREM1, BMP7, LDLRAD4, SMAD protein phosphorylation 9 6.34 1.31E-08 ENG pathway-restricted SMAD protein GDF3, SMAD7, GDF7, BMPR2, GDF10, GREM1, BMP7, LDLRAD4, phosphorylation -

The Role of Cdep in the Embryonic Morphogenesis of Drosophila Melanogaster
The Role of Cdep in the Embryonic Morphogenesis of Drosophila melanogaster Dissertation zur Erlangung des akademischen Grades Doctor rerum naturalium (Dr. rer.nat.) vorgelegt der Fakultät Mathematik und Naturwissenschaften der Technischen Universität Dresden von Anne Morbach Geboren am 05. August 1984 in Nordhausen, Deutschland Eingereicht am 30. Oktober 2015 Die Dissertation wurde in der Zeit von April 2012 bis Oktober 2015 im Max-Planck-Institut für molekulare Zellbiologie und Genetik angefertigt Gutachter: Prof. Dr. Elisabeth Knust Prof. Dr. Christian Dahmann Now, here, you see, it takes all the running you can do, to keep in the same place. Lewis Carroll, Through the Looking-Glass I Contents List of Figures V List of Tables VII Abstract IX List of Abbreviations XI 1 Introduction 1 1.1 Epithelial cell polarity . 1 1.1.1 Cellularization and formation of the primary epithelium . 1 1.1.1.1 Establishment of epithelial polarity and adhesion . 2 1.1.2 The epithelial polarity network . 3 1.1.3 Cell-cell adhesion . 5 1.1.3.1 Adherens junctions . 5 1.1.3.2 Septate junctions . 6 1.2 Epithelial movements in Drosophila embryonic morphogenesis . 6 1.2.1 Epithelial tube formation during Drosophila embryogenesis . 7 1.2.2 Coordinated migration of epithelial sheets during Dros. embryogenesis 7 1.2.2.1 FERM domain proteins in epithelial migration . 9 1.2.2.2 Cdep . 10 1.3 Mutagenesis with the CRISPR/Cas9 system . 12 2 Aim of My PhD Thesis Work 15 3 Preliminary Work 17 4 Results 19 4.1 A screen for novel regulators in Drosophila embryonic morphogenesis . -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Transcriptional Profiling Identifies the Lncrna PVT1 As a Novel Regulator of the Asthmatic Phenotype in Human Airway Smooth Muscle
Accepted Manuscript Transcriptional profiling identifies the lncRNA PVT1 as a novel regulator of the asthmatic phenotype in human airway smooth muscle Philip J. Austin, MSc, Eleni Tsitsiou, PhD, Charlotte Boardman, MD, Simon W. Jones, PhD, Mark A. Lindsay, PhD, Ian M. Adcock, PhD, Kian Fan Chung, MD PhD, Mark M. Perry, PhD PII: S0091-6749(16)30571-1 DOI: 10.1016/j.jaci.2016.06.014 Reference: YMAI 12203 To appear in: Journal of Allergy and Clinical Immunology Received Date: 5 April 2016 Revised Date: 24 May 2016 Accepted Date: 13 June 2016 Please cite this article as: Austin PJ, Tsitsiou E, Boardman C, Jones SW, Lindsay MA, Adcock IM, Chung KF, Perry MM, Transcriptional profiling identifies the lncRNA PVT1 as a novel regulator of the asthmatic phenotype in human airway smooth muscle, Journal of Allergy and Clinical Immunology (2016), doi: 10.1016/j.jaci.2016.06.014. This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain. ACCEPTED MANUSCRIPT 1 Transcriptional profiling identifies the lncRNA PVT1 as a novel 2 regulator of the asthmatic phenotype in human airway smooth muscle 3 4 Philip J. Austin MSc 1, Eleni Tsitsiou PhD 2, Charlotte Boardman MD 1, Simon W.