(12) Patent Application Publication (10) Pub. No.: US 2008/0044390 A1 Jin Et Al
Total Page:16
File Type:pdf, Size:1020Kb
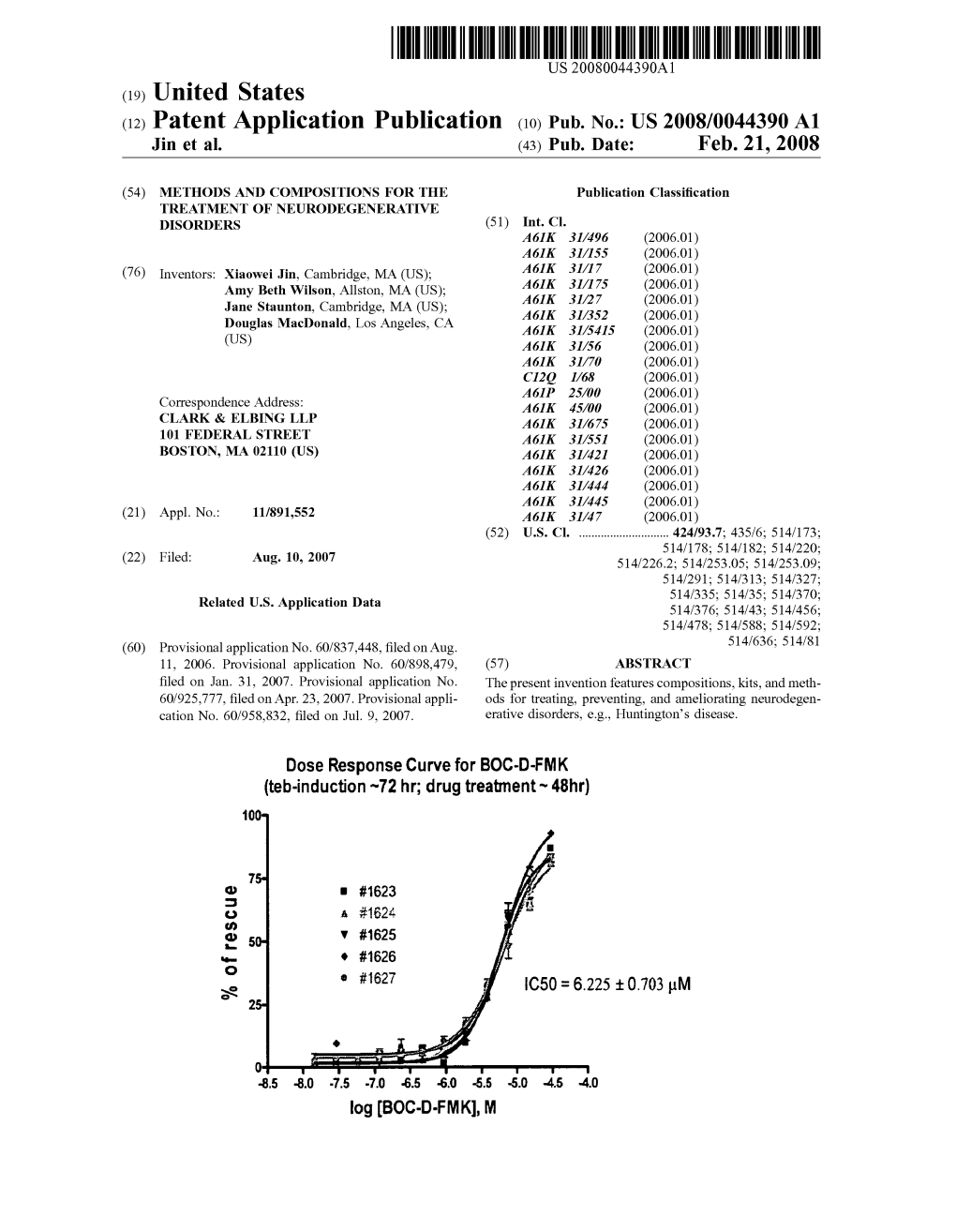
Load more
Recommended publications
-

Aldrich FT-IR Collection Edition I Library
Aldrich FT-IR Collection Edition I Library Library Listing – 10,505 spectra This library is the original FT-IR spectral collection from Aldrich. It includes a wide variety of pure chemical compounds found in the Aldrich Handbook of Fine Chemicals. The Aldrich Collection of FT-IR Spectra Edition I library contains spectra of 10,505 pure compounds and is a subset of the Aldrich Collection of FT-IR Spectra Edition II library. All spectra were acquired by Sigma-Aldrich Co. and were processed by Thermo Fisher Scientific. Eight smaller Aldrich Material Specific Sub-Libraries are also available. Aldrich FT-IR Collection Edition I Index Compound Name Index Compound Name 3515 ((1R)-(ENDO,ANTI))-(+)-3- 928 (+)-LIMONENE OXIDE, 97%, BROMOCAMPHOR-8- SULFONIC MIXTURE OF CIS AND TRANS ACID, AMMONIUM SALT 209 (+)-LONGIFOLENE, 98+% 1708 ((1R)-ENDO)-(+)-3- 2283 (+)-MURAMIC ACID HYDRATE, BROMOCAMPHOR, 98% 98% 3516 ((1S)-(ENDO,ANTI))-(-)-3- 2966 (+)-N,N'- BROMOCAMPHOR-8- SULFONIC DIALLYLTARTARDIAMIDE, 99+% ACID, AMMONIUM SALT 2976 (+)-N-ACETYLMURAMIC ACID, 644 ((1S)-ENDO)-(-)-BORNEOL, 99% 97% 9587 (+)-11ALPHA-HYDROXY-17ALPHA- 965 (+)-NOE-LACTOL DIMER, 99+% METHYLTESTOSTERONE 5127 (+)-P-BROMOTETRAMISOLE 9590 (+)-11ALPHA- OXALATE, 99% HYDROXYPROGESTERONE, 95% 661 (+)-P-MENTH-1-EN-9-OL, 97%, 9588 (+)-17-METHYLTESTOSTERONE, MIXTURE OF ISOMERS 99% 730 (+)-PERSEITOL 8681 (+)-2'-DEOXYURIDINE, 99+% 7913 (+)-PILOCARPINE 7591 (+)-2,3-O-ISOPROPYLIDENE-2,3- HYDROCHLORIDE, 99% DIHYDROXY- 1,4- 5844 (+)-RUTIN HYDRATE, 95% BIS(DIPHENYLPHOSPHINO)BUT 9571 (+)-STIGMASTANOL -

Ketamine As a Novel Treatment for Major Depressive Disorder and Bipolar Depression: a Systematic Review and Quantitative Meta-Analysis☆
General Hospital Psychiatry 37 (2015) 178–184 Contents lists available at ScienceDirect General Hospital Psychiatry journal homepage: http://www.ghpjournal.com Ketamine as a novel treatment for major depressive disorder and bipolar depression: a systematic review and quantitative meta-analysis☆ Ellen E. Lee, M.D., Megan P. Della Selva, M.D., Anson Liu, M.D., Seth Himelhoch, M.D., M.P.H. ⁎ Department of Psychiatry, University of Maryland School of Medicine, Baltimore, MD article info abstract Article history: Objective: Given the significant disability, morbidity and mortality associated with depression, the promising Received 9 September 2014 recent trials of ketamine highlight a novel intervention. A meta-analysis was conducted to assess the efficacy Revised 29 December 2014 of ketamine in comparison with placebo for the reduction of depressive symptoms in patients who meet criteria Accepted 8 January 2015 for a major depressive episode. Method: Two electronic databases were searched in September 2013 for English-language studies that were ran- Keywords: domized placebo-controlled trials of ketamine treatment for patients with major depressive disorder or bipolar Ketamine Depression depression and utilized a standardized rating scale. Studies including participants receiving electroconvulsive ther- Therapy apy and adolescent/child participants were excluded. Five studies were included in the quantitative meta-analysis. Meta-analysis Results: The quantitative meta-analysis showed that ketamine significantly reduced depressive symptoms. The overall effect size at day 1 was large and statistically significant with an overall standardized mean difference of 1.01 (95% confidence interval 0.69–1.34) (Pb.001), with the effects sustained at 7 days postinfusion. The heteroge- neity of the studies was low and not statistically significant, and the funnel plot showed no publication bias. -

Pureweigh®-FM
Manufacturers of Hypo-al ler gen ic Nutritional Sup ple ments PureWeigh®-FM INTRODUCED 2000 What Is It? than DHEA in stimulating the thermogenic enzymes of the liver, helping to support a leaner BMI (Body Mass PureWeigh®-FM is an encapsulated supplement companion Index) and healthy weight control. In a double blind to PureWeigh® PREMEAL Beverage containing banaba study involving 30 overweight adults, 7-KETO supported (Lagerstroemia speciosa L.) extract, green tea extract, healthy body composition and BMI when combined with taurine, 7-KETO™ DHEA, biotin, magnesium citrate and exercise.* chromium polynicotinate. PureWeigh®-FM may also be used independently of PureWeigh® PREMEAL Beverage to support • Biotin, facilitating protein, fat and carbohydrate healthy glucose metabolism and promote weight loss.* metabolism by acting as a coenzyme for numerous metabolic reactions. A clinical study reported that high Features Include dose administration of biotin helped promote healthy glucose metabolism. A number of animal studies support • Banaba extract, containing a triterpenoid compound this claim. Biotin may also act to promote transcription called corosolic acid, reported in studies to support and translation of glucokinase, an enzyme found in the healthy glucose function and absorption. A recent liver and pancreas that participates in the metabolism phase II, double-blind, placebo-controlled multi-center of glucose to form glycogen. In addition, a double-blind trial in Japan suggested that banaba extract maintained study reported that biotin supplementation may promote healthy glucose function and was well tolerated by healthy lipid metabolism, citing an inverse relationship volunteers. Furthermore, an independent U.S. between plasma biotin and total lipids.* preliminary clinical study reported statistically significant weight loss in human volunteers • Magnesium citrate, providing a highly bioavailable supplementing with a 1% corosolic acid banaba extract. -

Campro Catalog Stable Isotope
Introduction & Welcome Dear Valued Customer, We are pleased to present to you our Stable Isotopes Catalog which contains more than three thousand (3000) high quality labeled compounds. You will find new additions that are beneficial for your research. Campro Scientific is proud to work together with Isotec, Inc. for the distribution and marketing of their stable isotopes. We have been working with Isotec for more than twenty years and know that their products meet the highest standard. Campro Scientific was founded in 1981 and we provide services to some of the most prestigious universities, research institutes and laboratories throughout Europe. We are a research-oriented company specialized in supporting the requirements of the scientific community. We are the exclusive distributor of some of the world’s leading producers of research chemicals, radioisotopes, stable isotopes and environmental standards. We understand the requirements of our customers, and work every day to fulfill them. In working with us you are guaranteed to receive: - Excellent customer service - High quality products - Dependable service - Efficient distribution The highly educated staff at Campro’s headquarters and sales office is ready to assist you with your questions and product requirements. Feel free to call us at any time. Sincerely, Dr. Ahmad Rajabi General Manager 180/280 = unlabeled 185/285 = 15N labeled 181/281 = double labeled (13C+15N, 13C+D, 15N+18O etc.) 186/286 = 12C labeled 182/282 = d labeled 187/287 = 17O labeled 183/283 = 13C labeleld 188/288 = 18O labeled 184/284 = 16O labeled, 14N labeled 189/289 = Noble Gases Table of Contents Ordering Information.................................................................................................. page 4 - 5 Packaging Information .............................................................................................. -
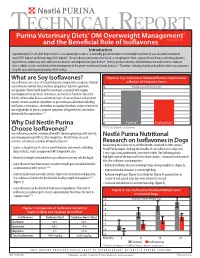
What Are Soy Isoflavones? Figure 2: Soy Isoflavones Reduced Plasma Isoprostanes Soy Isoflavones Are a Class of Natural Bioactive Compounds in Soybeans
TECHNICAL REPORT Purina Veterinary Diets® OM Overweight Management® brand canine dry formula and the Beneficial Role of Isoflavones Introduction Approximately 35% of adult dogs in the U.S. are overweight or obese1. A markedly greater incidence of overweight and obesity was observed in neutered male (59% higher) and female dogs (40% higher)1. Chronic obesity can increase the risk of, or complications from, several chronic diseases including diabetes, hypertension, pulmonary and cardiovascular disease, and degenerative joint disease2. Obesity produces chronic, mild inflammation and increases oxidative stress, which, in turn, contributes to the development of the above-mentioned chronic diseases.3,4 Therefore, reducing obesity and oxidative stress can promote a long life span and improved quality of life in dogs. What are Soy Isoflavones? Figure 2: Soy Isoflavones Reduced Plasma Isoprostanes Soy isoflavones are a class of natural bioactive compounds in soybeans. Natural a Marker of Oxidative Stress soy isoflavones include three chemical compounds: daidzein, genistein, 5 Plasma Isoprostanes (ng/ml) and glycitein. Many health benefits have been associated with regular consumption of soy products. In humans, soy has been found to reduce the 4 risk of cardiovascular disease and certain types of cancer (breast and prostate cancer); relieve a number of problems in post menopausal women including 3 hot flashes, osteoporosis, and decline in cognitive function; reduce cholesterol and triglycerides in plasma; improve symptoms of hypertension; and reduce 2 abdominal fat accumulation.5,6,7 1 Why Did Nestlé Purina 0 Control Isoflavones* Choose Isoflavones? *P<0.01 for Control vs. Isoflavones Soy isoflavones provide a number of benefits for managing dogs with obesity, and reducing rebound effects after weight loss. -

Applications of in Silico Methods to Analyze the Toxicity and Estrogen T Receptor-Mediated Properties of Plant-Derived Phytochemicals ∗ K
Food and Chemical Toxicology 125 (2019) 361–369 Contents lists available at ScienceDirect Food and Chemical Toxicology journal homepage: www.elsevier.com/locate/foodchemtox Applications of in silico methods to analyze the toxicity and estrogen T receptor-mediated properties of plant-derived phytochemicals ∗ K. Kranthi Kumara, P. Yugandharb, B. Uma Devia, T. Siva Kumara, N. Savithrammab, P. Neerajaa, a Department of Zoology, Sri Venkateswara University, Tirupati, 517502, India b Department of Botany, Sri Venkateswara University, Tirupati, 517502, India ARTICLE INFO ABSTRACT Keywords: A myriad of phytochemicals may have potential to lead toxicity and endocrine disruption effects by interfering Phytochemicals with nuclear hormone receptors. In this examination, the toxicity and estrogen receptor−binding abilities of a QSAR modeling set of 2826 phytochemicals were evaluated. The endpoints mutagenicity, carcinogenicity (both CAESAR and ISS Toxicity models), developmental toxicity, skin sensitization and estrogen receptor relative binding affinity (ER_RBA) Nuclear hormone receptor binding were studied using the VEGA QSAR modeling package. Alongside the predictions, models were providing pos- Self−Organizing maps sible information for applicability domains and most similar compounds as similarity sets from their training Clustering and classification schemes sets. This information was subjected to perform the clustering and classification of chemicals using Self−Organizing Maps. The identified clusters and their respective indicators were considered as potential hotspot structures for the specified data set analysis. Molecular screening interpretations of models wereex- hibited accurate predictions. Moreover, the indication sets were defined significant clusters and cluster in- dicators with probable prediction labels (precision). Accordingly, developed QSAR models showed good pre- dictive abilities and robustness, which observed from applicability domains, representation spaces, clustering and classification schemes. -

Estroquench™ Hormone Specific Formulation™
PRODUCT DATA DOUGLAS LABORATORIES® 08/2014 1 EstroQuench™ Hormone Specific Formulation™ DESCRIPTION EstroQuench™ is a Hormone Specific Formulation™ of ingredients that have documented anti-aromatase activity as well as androgenic adaptogens which support the function of endogenous aromatase inhibitors. Collectively these herbs promote minimal production and function of estrogens, while promoting testosterone function, including optimal sexual function in both genders. This formulation is designed to quench excessive production of estrogens and aberrant functions of them while supporting optimal function of androgens by maintaining the health of androgen producing glands.† Hormone Specific Formulation™ provided by Douglas Laboratories® and formulated by Dr. Joseph J Collins is created to support the optimal function of specific hormones through the use of hormone specific adaptogens, hormone specific agonists and hormone specific functional mimetics. This formulation may be used as part of a hormone health program with dietary and nutrient support. In addition, this formulation may be used by clinicians as an adjuvant to support optimal hormone health in patients who have been prescribed bioidentical hormone therapies. FUNCTIONS Aromatase (a cytochrome P450 enzyme {CYP19}) is the enzyme that controls the conversion of androgens to estrogens. More specifically, aromatase is the enzyme responsible for catalyzing the biosynthesis of androstenedione into estrone, and the biosynthesis of testosterone to estradiol. Estrogens include the broad range of aromatized hormones created form androgens. The specific attribute of estrogens that separate them from progestogens, androgens and corticoids is that estrogens are the only aromatized steroid hormones. Estradiol, the most potent endogenous estrogen, is biosynthesized by aromatization from androgens by aromatase (which is also called estrogen synthase). -

Test Report Comprehensive Hormone Insights™
698814 COMPREHENSIVE HORMONE INSIGHTS™ TEST REPORT Dr. Maximus, N.D. E: [email protected] Date of Collection: P: 403-241-4500 Time of Collection: F: 403-241-4501 Date of Receipt: www.rmalab.com Reported On: CHI Accession: 698814 Healthcare Professional Patient Age: Dr. Maximus, N.D. Date of Birth: Gender: Male F: Relevant Medications Biometrics Curcumin Height (in) : 73 Weight (lb) : 180 BMI : 24 Waist (in) : 35 Hip (in) : 41 CHI Accession: 698814 SUMMARY HMUS01 How to read the graphs LEGEND: 50 66 Sex Steroid Hormones 50 66 Middle third of 33 33 84 reference population Hormone Start of 83 100 80 100 highest 16 Percentile Precursors 16 Percentile third of Sum of Androgens Sum of Estrogens 50 66 reference 0 0 population (T, DHT, α+β androstanediol) Listed in Interp Guide 33 84 End of 100 16 lowest Percentile00 50 66 50 66 third of 33 33 84 reference 0 population Patient’s percentile rank 81 100 95 100 compared to reference 16 Percentile 16 Percentile population (see summary) DHEA + Metabolites Sum of Progesterone Metabolites 0 (DHEA + A + E) 0 α+β Pregnanediol Cortisol Melatonin Oxidative Stress Free Cortisol Profile (ng/mg) 100 50 66 50 66 33 84 33 84 80 64 100 0 100 16 Percentile 16 Percentile 60 6-sulfatoxy 8-Hydroxy-2- 0 Melatonin 0 deoxyguanosine 40 (Overnight) (Overnight) 20 6-sulfatoxymelatonin provides 8-hydroxy-2-deoxyguanosine is Cortisol/Creatinine (ng/mg) insight into melatonin levels. a marker of oxidative stress 0 Morning Dinner Bedtime A B C 50 66 Free cortisol Cortisol Metabolites 33 84 profile is used to provides a general Testosterone Cortisol assess diurnal assessment of 16 100 cortisol rhythm adrenal cortisol 16 Percentile Cortisol production Cortisol Metabolites 0 (α+β THF + THE) Testosterone Cortisol/Testosterone provides insight into relative catabolic (cortisol) and anabolic (testosterone) states. -

Aldrich Raman
Aldrich Raman Library Listing – 14,033 spectra This library represents the most comprehensive collection of FT-Raman spectral references available. It contains many common chemicals found in the Aldrich Handbook of Fine Chemicals. To create the Aldrich Raman Condensed Phase Library, 14,033 compounds found in the Aldrich Collection of FT-IR Spectra Edition II Library were excited with an Nd:YVO4 laser (1064 nm) using laser powers between 400 - 600 mW, measured at the sample. A Thermo FT-Raman spectrometer (with a Ge detector) was used to collect the Raman spectra. The spectra were saved in Raman Shift format. Aldrich Raman Index Compound Name Index Compound Name 4803 ((1R)-(ENDO,ANTI))-(+)-3- 4246 (+)-3-ISOPROPYL-7A- BROMOCAMPHOR-8- SULFONIC METHYLTETRAHYDRO- ACID, AMMONIUM SALT PYRROLO(2,1-B)OXAZOL-5(6H)- 2207 ((1R)-ENDO)-(+)-3- ONE, BROMOCAMPHOR, 98% 12568 (+)-4-CHOLESTEN-3-ONE, 98% 4804 ((1S)-(ENDO,ANTI))-(-)-3- 3774 (+)-5,6-O-CYCLOHEXYLIDENE-L- BROMOCAMPHOR-8- SULFONIC ASCORBIC ACID, 98% ACID, AMMONIUM SALT 11632 (+)-5-BROMO-2'-DEOXYURIDINE, 2208 ((1S)-ENDO)-(-)-3- 97% BROMOCAMPHOR, 98% 11634 (+)-5-FLUORODEOXYURIDINE, 769 ((1S)-ENDO)-(-)-BORNEOL, 99% 98+% 13454 ((2S,3S)-(+)- 11633 (+)-5-IODO-2'-DEOXYURIDINE, 98% BIS(DIPHENYLPHOSPHINO)- 4228 (+)-6-AMINOPENICILLANIC ACID, BUTANE)(N3-ALLYL)PD(II) CL04, 96% 97 8167 (+)-6-METHOXY-ALPHA-METHYL- 10297 ((3- 2- NAPHTHALENEACETIC ACID, DIMETHYLAMINO)PROPYL)TRIPH 98% ENYL- PHOSPHONIUM BROMIDE, 12586 (+)-ANDROSTA-1,4-DIENE-3,17- 99% DIONE, 98% 13458 ((R)-(+)-2,2'- 963 (+)-ARABINOGALACTAN BIS(DIPHENYLPHOSPHINO)-1,1'- -

Daidzein and Genistein Content of Cereals
European Journal of Clinical Nutrition (2002) 56, 961–966 ß 2002 Nature Publishing Group All rights reserved 0954–3007/02 $25.00 www.nature.com/ejcn ORIGINAL COMMUNICATION Daidzein and genistein content of cereals J Liggins1, A Mulligan1,2, S Runswick1 and SA Bingham1,2* 1Medical Research Council Dunn Human Nutrition Unit, Hills Road, Cambridge, UK; and 2European Prospective Investigation of Cancer, University of Cambridge, Cambridge, UK Objective: To analyse 75 cereals and three soy flours commonly eaten in Europe for the phytoestrogens daidzein and genistein. Design: The phytoestrogens daidzein and genistein were extracted from dried foods, and the two isoflavones quantified after hydrolytic removal of any conjugated carbohydrate. Completeness of extraction and any procedural losses of the isoflavones 0 0 were accounted for using synthetic daidzin (7-O-glucosyl-4 -hydroxyisoflavone) and genistin (7-O-glucosyl-4 5-dihydroxyiso- flavone) as internal standards. Setting: Foods from the Cambridge UK area were purchased, prepared for eating, which included cooking if necessary, and freeze dried. Three stock soy flours were also analysed. Results: Eighteen of the foods assayed contained trace or no detectable daidzein or genistein. The soy flours were rich sources, containing 1639 – 2117 mg=kg. The concentration of the two isoflavones in the remaining foods ranged from 33 to 11 873 mg=kg. Conclusion: These analyses will supply useful information to investigators determining the intake of phytoestrogens in cereal products in order to relate intakes to potential biological activities. Sponsorship: This work was supported by the United Kingdom Medical Research Council, Ministry of Agriculture Fisheries and Food (contract FS2034) and the United States of America Army (contract DAMD 17-97-1-7028). -

2018 Apigenin Male Infertility (2).Pdf
Original Article Thai Journal of Pharmaceutical Sciences Apigenin and baicalin, each alone or in low-dose combination, attenuated chloroquine induced male infertility in adult rats Amira Akilah, Mohamed Balaha*, Mohamed-Nabeih Abd-El Rahman, Sabiha Hedya Department of Pharmacology, Faculty of Medicine, Tanta University, Postal No. 31527, El-Gish Street, Tanta, Egypt Corresponding Author: Department of Pharmacology, ABSTRACT Faculty of Medicine, Tanta University, Postal No. 31527, Introduction: Male infertility is a worldwide health problem, which accounts for about 50% of all El-Gish Street, Tanta, Egypt. cases of infertility and considered as the most common single defined cause of infertility. Recently, Tel.: +201284451952. E-mail: apigenin and baicalin exhibited a powerful antioxidant and antiapoptotic activities. Consequently, in [email protected]. the present study, we evaluated the possible protective effect of apigenin and baicalin, either alone or Edu.Eg (M. Balaha). in low-dose combination, on a rat model of male infertility, regarding for its effects on the hormonal assay, testicular weight, sperm parameters, oxidative-stress state, apoptosis, and histopathological Received: Mar 06, 2018 changes. Material and methods: 12-week-old adult male Wister rats received 10 mg/kg/d Accepted: May 08, 2018 chloroquine orally for 30 days to induce male infertility. Either apigenin (30 or 15 mg/kg/d), baicalin Published: July 10, 2018 (100 or 50 mg/kg/d) or a combination of 15 mg/kg/d apigenin and 50 mg/kg/d baicalin received daily -

Download Product Insert (PDF)
PRODUCT INFORMATION (−)-Epigallocatechin Gallate Item No. 70935 CAS Registry No.: 989-51-5 Formal Name: 3,4-dihydro-5,7-dihydroxy-2R-(3,4,5- OH trihydroxyphenyl)-2H-1-benzopyran-3R- OH yl-3,4,5-trihydroxy-benzoate H HO O Synonym: EGCG OH MF: C22H18O11 O FW: 458.4 H OH OH Purity: ≥96% O UV/Vis.: λmax: 276 nm Supplied as: A crystalline solid OH Storage: -20°C OH Stability: ≥2 years Item Origin: Plant/Folium camelliae Information represents the product specifications. Batch specific analytical results are provided on each certificate of analysis. Laboratory Procedures (−)-Epigallocatechin gallate (EGCG) is supplied as a crystalline solid. A stock solution may be made by dissolving the EGCG in an organic solvent purged with an inert gas. EGCG is soluble in organic solvents such as ethanol, DMSO, and dimethyl formamide. The solubility of EGCG in these solvents is approximately 20, 25, and 30 mg/ml, respectively. Further dilutions of the stock solution into aqueous buffers or isotonic saline should be made prior to performing biological experiments. Ensure that the residual amount of organic solvent is insignificant, since organic solvents may have physiological effects at low concentrations. Organic solvent-free aqueous solutions of EGCG can be prepared by directly dissolving the crystalline compound in aqueous buffers. The solubility of EGCG in PBS (pH 7.2) is approximately 25 mg/ml. We do not recommend storing the aqueous solution for more than one day. Description EGCG is a phenol that has been found in green and black tea plants and has diverse biological activities.1-7 1 It is lytic against T.