On the Analytical Mechanics of Chemical Reactions. Classical Mechanics of Linear Collisions*
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

On the History of the Radiation Reaction1 Kirk T
On the History of the Radiation Reaction1 Kirk T. McDonald Joseph Henry Laboratories, Princeton University, Princeton, NJ 08544 (May 6, 2017; updated March 18, 2020) 1 Introduction Apparently, Kepler considered the pointing of comets’ tails away from the Sun as evidence for radiation pressure of light [2].2 Following Newton’s third law (see p. 83 of [3]), one might suppose there to be a reaction of the comet back on the incident light. However, this theme lay largely dormant until Poincar´e (1891) [37, 41] and Planck (1896) [46] discussed the effect of “radiation damping” on an oscillating electric charge that emits electromagnetic radiation. Already in 1892, Lorentz [38] had considered the self force on an extended, accelerated charge e, finding that for low velocity v this force has the approximate form (in Gaussian units, where c is the speed of light in vacuum), independent of the radius of the charge, 3e2 d2v 2e2v¨ F = = . (v c). (1) self 3c3 dt2 3c3 Lorentz made no connection at the time between this force and radiation, which connection rather was first made by Planck [46], who considered that there should be a damping force on an accelerated charge in reaction to its radiation, and by a clever transformation arrived at a “radiation-damping” force identical to eq. (1). Today, Lorentz is often credited with identifying eq. (1) as the “radiation-reaction force”, and the contribution of Planck is seldom acknowledged. This note attempts to review the history of thoughts on the “radiation reaction”, which seems to be in conflict with the brief discussions in many papers and “textbooks”.3 2 What is “Radiation”? The “radiation reaction” would seem to be a reaction to “radiation”, but the concept of “radiation” is remarkably poorly defined in the literature. -

Classical Mechanics
Classical Mechanics Hyoungsoon Choi Spring, 2014 Contents 1 Introduction4 1.1 Kinematics and Kinetics . .5 1.2 Kinematics: Watching Wallace and Gromit ............6 1.3 Inertia and Inertial Frame . .8 2 Newton's Laws of Motion 10 2.1 The First Law: The Law of Inertia . 10 2.2 The Second Law: The Equation of Motion . 11 2.3 The Third Law: The Law of Action and Reaction . 12 3 Laws of Conservation 14 3.1 Conservation of Momentum . 14 3.2 Conservation of Angular Momentum . 15 3.3 Conservation of Energy . 17 3.3.1 Kinetic energy . 17 3.3.2 Potential energy . 18 3.3.3 Mechanical energy conservation . 19 4 Solving Equation of Motions 20 4.1 Force-Free Motion . 21 4.2 Constant Force Motion . 22 4.2.1 Constant force motion in one dimension . 22 4.2.2 Constant force motion in two dimensions . 23 4.3 Varying Force Motion . 25 4.3.1 Drag force . 25 4.3.2 Harmonic oscillator . 29 5 Lagrangian Mechanics 30 5.1 Configuration Space . 30 5.2 Lagrangian Equations of Motion . 32 5.3 Generalized Coordinates . 34 5.4 Lagrangian Mechanics . 36 5.5 D'Alembert's Principle . 37 5.6 Conjugate Variables . 39 1 CONTENTS 2 6 Hamiltonian Mechanics 40 6.1 Legendre Transformation: From Lagrangian to Hamiltonian . 40 6.2 Hamilton's Equations . 41 6.3 Configuration Space and Phase Space . 43 6.4 Hamiltonian and Energy . 45 7 Central Force Motion 47 7.1 Conservation Laws in Central Force Field . 47 7.2 The Path Equation . -

Power Sources Challenge
POWER SOURCES CHALLENGE FUSION PHYSICS! A CLEAN ENERGY Summary: What if we could harness the power of the Sun for energy here on Fusion reactions occur when two nuclei come together to form one Earth? What would it take to accomplish this feat? Is it possible? atom. The reaction that happens in the sun fuses two Hydrogen atoms together to produce Helium. It looks like this in a very simplified way: Many researchers including our Department of Energy scientists and H + H He + ENERGY. This energy can be calculated by the famous engineers are taking on this challenge! In fact, there is one DOE Laboratory Einstein equation, E = mc2. devoted to fusion physics and is committed to being at the forefront of the science of magnetic fusion energy. Each of the colliding hydrogen atoms is a different isotope of In order to understand a little more about fusion energy, you will learn about hydrogen, one deuterium and one the atom and how reactions at the atomic level produce energy. tritium. The difference in these isotopes is simply one neutron. Background: It all starts with plasma! If you need to learn more about plasma Deuterium has one proton and one physics, visit the Power Sources Challenge plasma activities. neutron, tritium has one proton and two neutrons. Look at the The Fusion Reaction that happens in the SUN looks like this: illustration—do you see how the mass of the products is less than the mass of the reactants? That is called a mass deficit and that difference in mass is converted into energy. -

Physics 101 Today Chapter 5: Newton's Third
Physics 101 Today Chapter 5: Newton’s Third Law First, let’s clarify notion of a force : Previously defined force as a push or pull. Better to think of force as an interaction between two objects. You can’t push anything without it pushing back on you ! Whenever one object exerts a force on a second object, the second object exerts an equal and opposite force on the first. Newton’s 3 rd Law - often called “action-reaction ” Eg. Leaning against a wall. You push against the wall. The wall is also pushing on you, equally hard – normal/support force. Now place a piece of paper between the wall and hand. Push on it – it doesn’t accelerate must be zero net force. The wall is pushing equally as hard (normal force) on the paper in the opposite direction to your hand, resulting in zero Fnet . This is more evident when hold a balloon against the wall – it is squashed on both sides. Eg. You pull on a cart. It accelerates. The cart pulls back on you (you feel the rope get tighter). Can call your pull the “ action ” and cart’s pull the “ reaction ”. Or, the other way around. • Newton’s 3 rd law means that forces always come in action -reaction pairs . It doesn’t matter which is called the action and which is called the reaction. • Note: Action-reaction pairs never act on the same object Examples of action-reaction force pairs In fact it is the road’s push that makes the car go forward. Same when we walk – push back on floor, floor pushes us forward. -

HW 6, Question 1: Forces in a Rotating Frame
HW 6, Question 1: Forces in a Rotating Frame Consider a solid platform, like a \merry-go round", that rotates with respect to an inertial frame of reference. It is oriented in the xy plane such that the axis of rotation is in the z-direction, i.e. ! = !z^, which we can take to point out of the page. Now, let us also consider a person walking on the rotating platform, such that this person is walking radially outward on the platform with respect to their (non-inertial) reference frame with velocity v = vr^. In what follows for Question 1, directions in the non-inertial reference frame (i.e. the frame of the walking person) are defined in the following manner: the x0-direction points directly outward from the center of the merry-go round; the y0 points in the tangential direction of rotation in the plane of the platform; and the z0-direction points out of the page, and in the same direction as the z-direction for the inertial reference frame. (a) Identify all forces. Since the person's reference frame is non-inertial, we need to account for the various fictitious forces given the description above, as well as any other external/reaction forces. The angular rate of rotation is constant, and so the Euler force Feul = m(!_ × r) = ~0. The significant forces acting on the person are: 0 • the force due to gravity, Fg = mg = mg(−z^ ), 2 0 • the (fictitious) centrifugal force, Fcen = m! × (! × r) = m! rx^ , 0 • the (fictitious) Coriolis force, Fcor = 2mv × ! = 2m!v(−y^ ), • the normal force the person experiences from the rotating platform N = Nz^0, and • a friction force acting on the person, f, in the x0y0 plane. -

A Short History of Physics (Pdf)
A Short History of Physics Bernd A. Berg Florida State University PHY 1090 FSU August 28, 2012. References: Most of the following is copied from Wikepedia. Bernd Berg () History Physics FSU August 28, 2012. 1 / 25 Introduction Philosophy and Religion aim at Fundamental Truths. It is my believe that the secured part of this is in Physics. This happend by Trial and Error over more than 2,500 years and became systematic Theory and Observation only in the last 500 years. This talk collects important events of this time period and attaches them to the names of some people. I can only give an inadequate presentation of the complex process of scientific progress. The hope is that the flavor get over. Bernd Berg () History Physics FSU August 28, 2012. 2 / 25 Physics From Acient Greek: \Nature". Broadly, it is the general analysis of nature, conducted in order to understand how the universe behaves. The universe is commonly defined as the totality of everything that exists or is known to exist. In many ways, physics stems from acient greek philosophy and was known as \natural philosophy" until the late 18th century. Bernd Berg () History Physics FSU August 28, 2012. 3 / 25 Ancient Physics: Remarkable people and ideas. Pythagoras (ca. 570{490 BC): a2 + b2 = c2 for rectangular triangle: Leucippus (early 5th century BC) opposed the idea of direct devine intervention in the universe. He and his student Democritus were the first to develop a theory of atomism. Plato (424/424{348/347) is said that to have disliked Democritus so much, that he wished his books burned. -
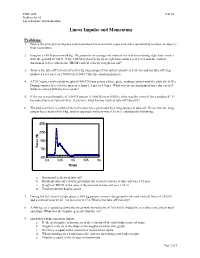
Linear Impulse and Momentum
EXSC 408L Fall '03 Problem Set #5 Linear Impulse and Momentum Linear Impulse and Momentum Problems: 1. Derive the principal of impulse and momentum from an initial cause and effect relationship to show an object’s final momentum. 2. Imagine a 100 N person walking. He generates an average net vertical reaction force during right foot contact with the ground of 100 N. If the TBCM vertical velocity at right foot contact is -0.2 m/s and the contact duration is 0.25 s, what is the TBCM vertical velocity at right toe off? 3. What is the take-off velocity of a 68.0 kg long jumper if his initial velocity is 8.50 m/s and his take-off step produces a net force of 2700 N for 0.200 s? Use the equation from #1. 4. A USC runner with a body weight of 490.5 N runs across a force plate, making contact with the plate for 0.25 s. During contact, her velocity increases from 2.5 m/s to 4.0 m/s. What was the net horizontal force she exerted while in contact with the force plate? 5. If the net vertical impulse of a 580 N person is 1800 Ns over 0.500 s, what was the vertical force produced? If her initial vertical velocity was –0.200 m/s, what was her vertical take-off velocity? 6. The plot seen here is a plot of the horizontal force generated by a long-jumper at take-off. Given that the long- jumper has a mass of 68.0 kg, and his approach velocity was 8.30 m/s, calculate the following: 3750 2750 1750 Force (N) 750 -250 0.4 0.45 0.5 0.55 0.6 Time(s) a. -

Newton's Third Law (Lecture 7) Example the Bouncing Ball You Can Move
3rd Law Newton’s third law (lecture 7) • If object A exerts a force on object B, then object B exerts an equal force on object A For every action there is an in the opposite direction. equal and opposite reaction. B discuss collisions, impulse, A momentum and how airbags work B Æ A A Æ B Example The bouncing ball • What keeps the box on the table if gravity • Why does the ball is pulling it down? bounce? • The table exerts an • It exerts a downward equal and opposite force on ground force upward that • the ground exerts an balances the weight upward force on it of the box that makes it bounce • If the table was flimsy or the box really heavy, it would fall! Action/reaction forces always You can move the earth! act on different objects • The earth exerts a force on you • you exert an equal force on the earth • The resulting accelerations are • A man tries to get the donkey to pull the cart but not the same the donkey has the following argument: •F = - F on earth on you • Why should I even try? No matter how hard I •MEaE = myou ayou pull on the cart, the cart always pulls back with an equal force, so I can never move it. 1 Friction is essential to movement You can’t walk without friction The tires push back on the road and the road pushes the tires forward. If the road is icy, the friction force You push on backward on the ground and the between the tires and road is reduced. -

A Cultural History of Physics
Károly Simonyi A Cultural History of Physics Translated by David Kramer Originally published in Hungarian as A fizika kultûrtörténete, Fourth Edition, Akadémiai Kiadó, Budapest, 1998, and published in German as Kulturgeschichte der Physik, Third Edition, Verlag Harri Deutsch, Frankfurt am Main, 2001. First Hungarian edition 1978. CRC Press Taylor & Francis Group 6000 Broken Sound Parkway NW, Suite 300 Boca Raton, FL 33487-2742 © 2012 by Taylor & Francis Group, LLC CRC Press is an imprint of Taylor & Francis Group, an Informa business No claim to original U.S. Government works Printed in the United States of America on acid-free paper Version Date: 20111110 International Standard Book Number: 978-1-56881-329-5 (Hardback) This book contains information obtained from authentic and highly regarded sources. Reasonable efforts have been made to publish reliable data and information, but the author and publisher cannot assume responsibility for the validity of all materials or the consequences of their use. The authors and publishers have attempted to trace the copyright holders of all material reproduced in this publication and apologize to copyright holders if permission to publish in this form has not been obtained. If any copyright material has not been acknowl- edged please write and let us know so we may rectify in any future reprint. Except as permitted under U.S. Copyright Law, no part of this book may be reprinted, reproduced, transmitted, or utilized in any form by any electronic, mechanical, or other means, now known or hereafter invented, including photocopying, microfilming, and recording, or in any information storage or retrieval system, without written permission from the publishers. -

Analysis of Standing Vertical Jumps Using a Force Platform Nicholas P
Analysis of standing vertical jumps using a force platform Nicholas P. Linthornea) School of Exercise and Sport Science, The University of Sydney, Sydney, New South Wales, Australia ͑Received 9 March 2001; accepted 8 May 2001͒ A force platform analysis of vertical jumping provides an engaging demonstration of the kinematics and dynamics of one-dimensional motion. The height of the jump may be calculated ͑1͒ from the flight time of the jump, ͑2͒ by applying the impulse–momentum theorem to the force–time curve, and ͑3͒ by applying the work–energy theorem to the force-displacement curve. © 2001 American Association of Physics Teachers. ͓DOI: 10.1119/1.1397460͔ I. INTRODUCTION subject. In terrestrial human movement, the force exerted by the platform on the body is commonly called the ‘‘ground A force platform can be an excellent teaching aid in un- reaction force.’’ dergraduate physics classes and laboratories. Recently, The jumps discussed in this article we recorded using a Cross1 showed how to increase student interest and under- Kistler force platform that was set in concrete in the floor of standing of elementary mechanics by using a force platform our teaching laboratory.3 The vertical component of the to study everyday human movement such as walking, run- ground reaction force of the jumper was sampled at 1000 Hz ning, and jumping. My experiences with using a force plat- and recorded by an IBM compatible PC with a Windows 95 form in undergraduate classes have also been highly favor- operating system. Data acquisition and analysis of the jumps able. The aim of this article is to show how a force platform were performed using a custom computer program ͑JUMP analysis of the standing vertical jump may be used in teach- ANALYSIS͒ that was written using LABVIEW virtual instru- ing the kinematics and dynamics of one-dimensional motion. -

The Amazing Normal Forces
THE AMAZING NORMAL FORCES Horia I. Petrache Department of Physics, Indiana University Purdue University Indianapolis Indianapolis, IN 46202 November 9, 2012 Abstract This manuscript is written for students in introductory physics classes to address some of the common difficulties and misconceptions of the normal force, especially the relationship between normal and friction forces. Accordingly, it is intentionally informal and conversational in tone to teach students how to build an intuition to complement mathematical formalism. This is accomplished by beginning with common and everyday experience and then guiding students toward two realizations: (i) That real objects are deformable even when deformations are not easily visible, and (ii) that the relation between friction and normal forces follows from the action-reaction principle. The traditional formulae under static and kinetic conditions are then analyzed to show that peculiarity of the normal-friction relationship follows readily from observations and knowledge of physics principles. 1 1. Normal forces: amazing or amusing? Learning about normal forces can be a life changing event. In introductory physics, we accept and embrace these totally mysterious things. Suddenly, normal forces become a convenient answer to everything: they hold objects on floors, on walls, in elevators and even on ceilings. They lift heavy weights on platforms, let footballs bounce, basketball players jump, and as if this was not enough, they even tell friction what to do. (Ah, the amazing friction forces – yet another amazing story! [1]) Life before physics becomes inexplicable. This article is about building an intuition about normal forces using the action-reaction law of mechanics and the fact that real objects are deformable. -

Proton Transfer Reaction Mass Spectrometry (Ptr-Ms)
28 PROTON TRANSFER REACTION MASS SPECTROMETRY ( PTR - MS ) Yujie Wang , Chengyin Shen , Jianquan Li , Haihe Jiang , and Yannan Chu 28.1 INTRODUCTION is measured. If a chromatographic separation method is not used prior to MS, then the resulting mass spectra Proton transfer reaction mass spectrometry ( PTR - MS ) from EI may be so complicated that identifi cation and was fi rst developed at the Institute of Ion Physics of quantifi cation of the compounds can be very diffi cult. In Innsbruck University in the 1990s. Nowadays, PTR - MS PTR - MS instrument, the hollow cathode discharge is is a well - developed and commercially available tech- served as a typical ion source [1] , although plane elec- nique for the on - line monitoring of trace volatile organic trode direct current discharge [2] and radioactive ion- compound s ( VOC s) down to parts per trillion by volume ization sources [3] recently have been reported. All of (ppt) level. PTR - MS has some advantages such as rapid the ion sources are used to generate clean and intense + response, soft chemical ionization ( CI ), absolute quan- primary reagent ions like H3 O . Water vapor is a regular tifi cation, and high sensitivity. In general, a standard gas in the hollow cathode discharge where H2 O mole- PTR - MS instrument consists of external ion source, cule can be ionized according to the following ways [4] : drift tube, and mass analysis detection system. Figure 28.1 illustrates the basic composition of the PTR - MS + ee+→++HO22 H O 2, (28.1) instrument constructed in our laboratory using a quad- +→+++ rupole mass spectrometer as the detection system.