Development of Acetylcholinesterase Biosensors for Harmful Substances Detection in Food and Environment
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Historical Perspectives on Apple Production: Fruit Tree Pest Management, Regulation and New Insecticidal Chemistries
Historical Perspectives on Apple Production: Fruit Tree Pest Management, Regulation and New Insecticidal Chemistries. Peter Jentsch Extension Associate Department of Entomology Cornell University's Hudson Valley Lab 3357 Rt. 9W; PO box 727 Highland, NY 12528 email: [email protected] Phone 845-691-7151 Mobile: 845-417-7465 http://www.nysaes.cornell.edu/ent/faculty/jentsch/ 2 Historical Perspectives on Fruit Production: Fruit Tree Pest Management, Regulation and New Chemistries. by Peter Jentsch I. Historical Use of Pesticides in Apple Production Overview of Apple Production and Pest Management Prior to 1940 Synthetic Pesticide Development and Use II. Influences Changing the Pest Management Profile in Apple Production Chemical Residues in Early Insect Management Historical Chemical Regulation Recent Regulation Developments Changing Pest Management Food Quality Protection Act of 1996 The Science Behind The Methodology Pesticide Revisions – Requirements For New Registrations III. Resistance of Insect Pests to Insecticides Resistance Pest Management Strategies IV. Reduced Risk Chemistries: New Modes of Action and the Insecticide Treadmill Fermentation Microbial Products Bt’s, Abamectins, Spinosads Juvenile Hormone Analogs Formamidines, Juvenile Hormone Analogs And Mimics Insect Growth Regulators Azadirachtin, Thiadiazine Neonicotinyls Major Reduced Risk Materials: Carboxamides, Carboxylic Acid Esters, Granulosis Viruses, Diphenyloxazolines, Insecticidal Soaps, Benzoyl Urea Growth Regulators, Tetronic Acids, Oxadiazenes , Particle Films, Phenoxypyrazoles, Pyridazinones, Spinosads, Tetrazines , Organotins, Quinolines. 3 I Historical Use of Pesticides in Apple Production Overview of Apple Production and Pest Management Prior to 1940 The apple has a rather ominous origin. Its inception is framed in the biblical text regarding the genesis of mankind. The backdrop appears to be the turbulent setting of what many scholars believe to be present day Iraq. -

Laboratory Test Report
Aromatic Plant Research Center 230 N 1200 E STE 100 Lehi, UT 84043 www.aromaticplant.org Laboratory Test Report SAMPLE NAME : Cypress CLIENT NAME : ACHS CLIENT LOT # : 36186C Column: ZB5 (60 m length × 0.25 mm inner diameter × 0.25 μm film thickness) Instrument: Shimadzu GCMS-QP2010 Ultra Carrier gas: Helium 80 psi Temperature ramp: 2 degrees Celsius per minute up to 260-degree Celsius Split ratio: 30:1 Sample preparation: 5% w/v solution with Dichloromethane Interpretation on sample: This analysis of Cypress essential oil meets the standard chemical profile of Cupressus sempervirens essential oil. Analyzed by: Dr. Prabodh Satyal Reviewed by: Ambika Poudel Issued Date: 9/25/2020 Copyright © 2020 by Aromatic Plant Research Center (APRC). All rights reserved. The information contained in this document may not be used, published or redistributed, including online, without the prior written consent of APRC. 1 / 4 24,426,457 Chromatogram Injection Volume Lot# Client Name Sample Name Sample Type Analyzed Analyzed by Sample Information www.aromaticplant.org UT Lehi, 84043 100 STE 230N E 1200 Center Aromatic Plant Research 10.856 Copyright © 2020 by Aromatic Plant Research Center (APRC). All rights reserved. Copyright information The All (APRC). rights reserved. this document contained in Center may © 2020by Aromatic Plantnot be used, published Research or 11.74611.898 13.37013.18113.088 12.429 14.335 14.41714.716 15.343 16.015 16.405 16.628 17.032 17.18317.501 18.00417.910 17.812 20.0 18.866 19.30119.646 21.153 21.744 21.445 22.392 : : : 0.30 : : : : ACHS Cy Esse 9/25/2020 7:12:23 AM AM 7:12:23 9/25/2020 Dr. -

4C Pesticide Lists
4C PESTICIDE LISTS Version 4.0 II 4C PESTICIDE LISTS Copyright notice © 2020 4C Services GmbH This document is protected by copyright. It is freely available from the 4C website or upon request. No part of this copyrighted document may be changed or amended. The document may not be duplicated or copied in any form or by any means for commercial purpose without permission of 4C Services. Document Title: 4C Pesticide Lists Version 4.0 Valid from: 01 July 2020 III Content List of Tables ........................................................................................................................ IV Abbreviations ....................................................................................................................... IV 4C PESTICIDE LISTS 1 Introduction ................................................................................................................... 5 2 Selection Criteria Used for the 4C Pesticide Lists .......................................................... 5 3 4C Red List Pesticides: 4C Code of Conduct Requirements and Actions to be Promoted .................................. 6 4 4C Yellow List Pesticides: 4C Code of Conduct Requirements and Actions to be Promoted .................................. 7 © 4C Services GmbH IV List of Tables Table 1: 4C list of unacceptable pesticides ............................................................................ 8 Table 2: 4C red pesticide list ................................................................................................. 9 Table -

Metabolism of Methiocarb and Carbaryl by Rat and Human Livers and Plasma, and Effect on Their PXR, CAR and Pparα Activities
The Journal of Toxicological Sciences (J. Toxicol. Sci.) 677 Vol.41, No.5, 677-691, 2016 Original Article Metabolism of methiocarb and carbaryl by rat and human livers and plasma, and effect on their PXR, CAR and PPARα activities Chieri Fujino1, Yuki Tamura2, Satoko Tange1, Hiroyuki Nakajima3,4, Seigo Sanoh1, Yoko Watanabe2, Naoto Uramaru2, Hiroyuki Kojima5, Kouichi Yoshinari3,4, Shigeru Ohta1 and Shigeyuki Kitamura2 1Graduate School of Biomedical and Health Sciences, Hiroshima University, Kasumi 1-2-3, Minami-ku, Hiroshima 734-8553, Japan 2Nihon Pharmaceutical University, Komuro 10281, Ina-machi, Kitaadachi-gun, Saitama 362-0806, Japan 3Graduate School of Pharmaceutical Sciences, Tohoku University, 6-3 Aramaki-Aoba, Aoba-ku, Sendai 980-8578, Japan 4School of Pharmaceutical Sciences, University of Shizuoka, 52-1 Yada, Suruga-ku, Shizuoka 422-8526, Japan 5Hokkaido Institute of Public Health, Kita-19, Nishi-12, Kita-ku, Sapporo 060-0819, Japan (Received April 14, 2016; Accepted August 2, 2016) ABSTRACT — The oxidative, reductive, and hydrolytic metabolism of methiocarb and the hydrolyt- ic metabolism of carbaryl by liver microsomes and plasma of rats or humans were examined. The effects of the metabolism of methiocarb and carbaryl on their nuclear receptor activities were also examined. When methiocarb was incubated with rat liver microsomes in the presence of NADPH, methiocarb sul- foxide, and a novel metabolite, methiocarb sulfone were detected. Methiocarb sulfoxide was oxidized to the sulfone by liver microsomes and reduced back to methiocarb by liver cytosol. Thus, the intercon- version between methiocarb and the sulfoxide was found to be a new metabolic pathway for methiocarb by liver microsomes. -

Lists for Pesticide Management
Rainforest Alliance Lists for Pesticide Management Lists of Prohibited and Risk Mitigation Use Pesticides July, 2017 Version 1.3 D.R. © 2017 Red de Agricultura Sostenible, A.C. This document is provided by Red de Agricultura Sostenible, A.C. (also known as Sustainable Agriculture Network) to Rainforest Alliance, Inc. and/or to its successors, under the terms and subject to the limitations set forth in the perpetual, exclusive, non-transferrable license granted by Red de Agricultura Sostenible, A.C. in favor of Rainforest Alliance, Inc., or its successors under the terms and conditions set forth in an agreement between the parties (the “Agreement”), in the understanding that: 1. All content of this document, including, but not limited to text, logos, if any, graphics, photographs, trade names, etc. of Red de Agricultura Sostenible, A.C, is subject to copyright protection in favor of the Red de Agricultura Sostenible, A.C. and third party owners who have duly authorized the inclusion of their work, under the provisions of the Mexican Federal Law on Copyright (Ley Federal del Derecho de Autor) and other related national and / or international laws. The Rainforest Alliance name and trademarks are the sole property of Rainforest Alliance. 2. Rainforest Alliance, Inc., and / or its successors, shall only use the copyrighted material under the terms and conditions of the Agreement. 3. Under no circumstance shall it be understood that a license, of any kind, over this document has been granted to any third party different from Rainforest Alliance, Inc., or its successors. 4. Except for the terms and conditions set forth in the Agreement, under no circumstance shall it be understood that Red de Agricultura Sostenible, A.C. -
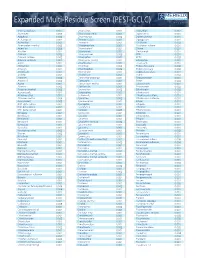
Pesticide Expanded Multi-Residue Screens
www.merieuxnutrisciences.com/us Expanded Multi-Residue Screen (PEST-GCLC) Tel: +1 (312) 938-5151 3-OH-carbofuran 0.001 Chloridazon 0.002 Dioxathion 0.002 Abamectin 0.005 Chlorimuron ethyl 0.001 Diphenamid 0.001 Acephate 0.002 Chlormephos 0.002 Diphenylamine 0.001 Acetamipred 0.002 Chlorobenzilate 0.001 Dipropetryn 0.001 Acetochlor 0.001 Chloroneb 0.001 Disulfoton 0.002 Acibenzolar-s-methyl 0.002 Chloropropilate 0.001 Disulfoton sulfone 0.001 Aclonifen 0.005 Chlorothalonil 0.001 Diuron 0.001 Alachlor 0.001 Chloroxuron 0.001 Dodemorph 0.001 Aldicarb 0.002 Chlorpropham 0.003 EPN 0.001 Aldicarb sulfone 0.002 Chlorpyrifos 0.001 EPTC 0.003 Aldicarb sulfoxide 0.002 Chlorpyrifos-methyl 0.001 Edifenphos 0.001 Aldrin 0.001 Chlorthiamid 0.002 Emamectin 0.001 Allidochlor 0.001 Chlorthion 0.002 Endosulfan alpha 0.002 Ametryn 0.001 Chlorthiophos 0.002 Endosulfan beta 0.003 Aminocarb 0.001 Chlortoluron 0.001 Endosulfan sulfate 0.001 Anilofos 0.001 Chlozolinate 0.001 Endrin 0.002 Aramite I 0.002 Clodinafop-propargyl 0.001 Epoxyconazole 0.001 Aramite II 0.003 Clomazone 0.001 Erbon 0.003 Aspon 0.001 Cloquintocet-methyl 0.001 Esfenvalerate 0.001 Atrazine 0.002 Clothianidin 0.001 Etaconazole 0.002 Atrazine-desethyl 0.002 Coumaphos 0.002 Ethaluralin 0.002 Azaconazole 0.001 Crotoxyphos 0.003 Ethiofencarb 0.001 Azinphos-ethyl 0.002 Cruformate 0.001 Ethiofencarb sulfone 0.001 Azinphos-methyl 0.006 Cyanazine 0.002 Ethiofencarb sulfoxide 0.001 Azoxystrobin 0.003 Cyanofenphos 0.001 Ethion 0.001 BHC alpha isomer 0.001 Cyanophos 0.001 Ethiprole 0.001 BHC beta isomer -

Recommended Classification of Pesticides by Hazard and Guidelines to Classification 2019 Theinternational Programme on Chemical Safety (IPCS) Was Established in 1980
The WHO Recommended Classi cation of Pesticides by Hazard and Guidelines to Classi cation 2019 cation Hazard of Pesticides by and Guidelines to Classi The WHO Recommended Classi The WHO Recommended Classi cation of Pesticides by Hazard and Guidelines to Classi cation 2019 The WHO Recommended Classification of Pesticides by Hazard and Guidelines to Classification 2019 TheInternational Programme on Chemical Safety (IPCS) was established in 1980. The overall objectives of the IPCS are to establish the scientific basis for assessment of the risk to human health and the environment from exposure to chemicals, through international peer review processes, as a prerequisite for the promotion of chemical safety, and to provide technical assistance in strengthening national capacities for the sound management of chemicals. This publication was developed in the IOMC context. The contents do not necessarily reflect the views or stated policies of individual IOMC Participating Organizations. The Inter-Organization Programme for the Sound Management of Chemicals (IOMC) was established in 1995 following recommendations made by the 1992 UN Conference on Environment and Development to strengthen cooperation and increase international coordination in the field of chemical safety. The Participating Organizations are: FAO, ILO, UNDP, UNEP, UNIDO, UNITAR, WHO, World Bank and OECD. The purpose of the IOMC is to promote coordination of the policies and activities pursued by the Participating Organizations, jointly or separately, to achieve the sound management of chemicals in relation to human health and the environment. WHO recommended classification of pesticides by hazard and guidelines to classification, 2019 edition ISBN 978-92-4-000566-2 (electronic version) ISBN 978-92-4-000567-9 (print version) ISSN 1684-1042 © World Health Organization 2020 Some rights reserved. -

1 of 3 GC+LC-USA
Updated: 07/18/2016 1 of 3 GC+LC-USA Limit of Quantitation (LOQ): 0.010 mg/kg (ppm) Sample Types: Low Fat Content Samples Minimum Sample Size: 100 grams (~1/4 pound). Certain products require more for better sample representation. Instrument: GC-MS/MS and LC-MS/MS Turnaround: 24-48 hours Accreditation: Part of AGQ USA's ISO/IEC 17025 Accreditation Scope 4,4'-Dichlorobenzophenone Bupirimate Cyantraniliprole Diflufenican Abamectin Buprofezin Cyazofamid Dimethoate Acephate Butachlor Cycloate Dimethoate (Sum) Acequinocyl Butocarboxim Cycloxydim Dimethomorph Acetamiprid Butralin Cyflufenamid Diniconazole Acetochlor Cadusafos Cyfluthrin Dinocap Acrinathrin Captafol Cymoxanil Dinotefuran Alachlor Captan Cyproconazole Diphenylamine Aldicarb Captan (Sum) Cyprodinil Disulfoton Aldicarb (Sum) Carbaryl Cyromazine Disulfoton (Sum) Aldicarb-sulfone Carbofuran DDD-o,p Disulfoton-sulfone Aldicarb-sulfoxide Carbofuran-3-hydroxy DDD-p,p +DDT-o,p Disulfoton-sulfoxide Aldrin Carbophenothion DDE-o,p Ditalimfos Ametryn Carbosulfan DDE-p,p Diuron Amitraz Carboxine DDT (Sum) Dodemorph Atrazine Carfentrazone-ethyl DDT-p,p Dodine Azadirachtin Chinomethionat DEET Emamectin Benzoate Azamethiphos Chlorantraniliprole Deltamethrin Endosulfan (A+B+Sulf) Azinphos-ethyl Chlordane Demeton Endosulfan Alfa Azinphos-methyl Chlordane Trans Demeton-S-methyl-sulfone Endosulfan Beta Azoxystrobin Chlorfenapyr Desmedipham Endosulfan Sulfate Benalaxyl Chlorfenson Diafenturion Endrin Ben-Carb-TPM (Sum) Chlorfenvinphos Dialifos EPN Bendiocarb Chlorfluazuron Diazinon Epoxiconazole -

Cumulative and Comprehensive Subject Matter Index Volumes 171–180
Cumulative and Comprehensive Subject Matter Index Volumes 171–180 Abamectin, avermectin B1, 171:112 Acanthamoeba, human infections caused, 1 Aaetnefcson crop pests/benefi- 180:116Abamectin,effects cial insects, 171:115 Acanthamoeba, in air, dust & soil, 180: 0 Aaetnevrnetleffects, 171: 103Abamectin,environmental 115 Acanthamoeba, in animal wastes, 180:102 Abamectin, non-target insect effects, 171: Acanthamoeba, in sewage & biosolids, 116 180:102 Abamectin, resistance induction insect Acanthamoeba, in surface waters, 180: pests, 171:116 100 Abbreviations, scientific organizations, Acanthamoeba, in swimming pools, spas, 172:119 180:102 Abbreviations, toxicological, 176:4 Acanthamoeba, in tapwater & bottled wa- Abiotic coupling, manganese oxidation of ter, 180:101 Cr(III), 178:121 Acanthamoeba infection in AIDS patients, Abiotic hydrolysis, pesticides aquatic envi- 180:114 ronment, 175:79 ff. Acanthamoeba, interactions with Legio- Abiotic pesticide hydrolysis, adsorption ef- nella pneumophila, 180:114 fects, 175:85 Acanthamoeba keratitis, contact lenses as- Abiotic pesticide hydrolysis, clay effects, sociation, 180:119 175:88 Acanthamoeba keratitis, diagnosis, 180: Abiotic pesticide hydrolysis, cosolvent ef- 106 fects, 175:85 Acanthamoeba keratitis, eye infections, Abiotic pesticide hydrolysis, dissolved or- 180:104 ganic matter effects, 175:86 Acanthamoeba keratitis, immunity, 180: Abiotic pesticide hydrolysis, metal ions/ 110 175:88 Acanthamoeba keratitis, incidence &oxideseffects, Abiotic pesticide hydrolysis, micelle ef- pathogenicity, -

Electroanalytical Study of the Pesticide Ethiofencarb
Electroanalytical Study of the Pesticide Ethiofencarb M. Fa´tima Barroso, M. Carmo V. F. Vaz, M. Goreti F. Sales, Paula Paı´ga, and Cristina Delerue-Matos Abstract: A detailed study of voltammetric behavior of ethiofencarb (ETF) is reported using glassy carbon electrode (GCE) and hanging mercury drop electrode (HMDE). With GCE, it is possible to verify that the oxidative mechanism is irreversible, indepen-dent of pH, and the maximum intensity current was observed at þ1.20 V vs. AgCl/Ag at pH 1.9. A linear calibration line was obtained from 1.0 Â 1024 to 8.0 Â 1024 mol L21 with SWV method. To complete the electrochemical knowledge of ETF pesticide, the reduction was also explored with HMDE. A well-defined peak was observed at –1.00 V vs. AgCl/Ag in a large range of pH with higher signal at pH 7.0. Linearity was obtained in 4.2 Â 1026 and 9.4 Â 1026 mol L21 ETF concentration range. An immediate alkaline hydrolysis of ETF was executed, producing a phenolic compound (2-ethylthiomethylphenol) (EMP), and the electrochemical activity of the product was examined. It was deduced that it is oxidized on GCE at þ0.75 V vs. AgCl/Ag with a maximum peak intensity current at pH 3.2, but the compound had no reduction activity on HMDE. Using the decrease of potential peak, a flow injection analysis (FIA) system was developed connected to an amperometric detector, enabling the determination of EMP over concentration range of 1.0 Â 1027 and 1.0 Â 1025 mol L21 at a sampling rate of 60 h21. -

Pesticide Data Program Annual Summary for Calendar Year 2018
United States Department of Agriculture December 2019 Dear Reader: We are pleased to present the Pesticide Data Program’s (PDP) 28th Annual Summary for calendar year 2018. The U.S. Department of Agriculture (USDA), Agricultural Marketing Service (AMS) conducts this program each year to collect data on pesticide residues in food. This report shows that when pesticide residues are found on foods, they are nearly always at levels below the tolerance, or maximum amount of a pesticide allowed to remain in or on a food, that is set by the U.S. Environmental Protection Agency (EPA). PDP provides high-quality, nationally representative data to help ensure consumer confidence in the foods they provide to their families. Over 99 percent of the products sampled through PDP had residues below the EPA tolerances. Ultimately, if EPA determines a pesticide is not safe for human consumption, it is removed from the market. The PDP tests a wide variety of domestic and imported foods, with a strong focus on foods that are consumed by infants and children. EPA relies on PDP data to conduct dietary risk assessments and to ensure that any pesticide residues in foods remain at safe levels. USDA uses the data to better understand the relationship of pesticide residues to agricultural practices and to enhance USDA’s Integrated Pest Management objectives. USDA also works with U.S. growers to improve agricultural practices. The PDP is not designed for enforcement of EPA pesticide residue tolerances. Rather, the U.S. Food and Drug Administration (FDA) is responsible for enforcing EPA tolerances. PDP provides FDA and EPA with monthly reports of pesticide residue testing and informs the FDA if residues detected exceed the EPA tolerance or have no EPA tolerance established. -

PDP Pesticide History Years Each Pesticide Was Reported from 1991 - 2020
USDA, AMS, S&T, MPD - Pesticide Data Program (PDP) 2 November 2020 PDP Pesticide History Years each pesticide was reported from 1991 - 2020 Pest Pesticide Name Code Years Pesticide was Reported 1,2,4-Triazole A68 2003 - 2007 1-Naphthol 382 1994 - 2000, 2003 - 2020 2,3,5-Trimethacarb A72 2020 2,4,5-T 312 2001 - 2016 2,4,5-TP AJE 2010 - 2013 2,4-D 026 1992 - 1998, 2002 - 2020 2,4-DB 317 1996 - 1997, 2003 - 2016, 2019 2,4-dimethyl aniline (2,4 DMA) AGQ 2007 - 2008 2,4-dimethylphenyl formamide (2,4-DMPF) AGR 2007 - 2012, 2016 - 2020 2,6-dichlorobenzamide 272 2010, 2017 2,6-DIPN AFZ 2016 - 2020 3,5-Dichloroaniline ABM 2003 3-Hydroxycarbofuran 512 1993 - 2020 3-ketocarbofuran 218 2001 - 2002 4,4-dibromobenzophenone AGS 2007 - 2008 4-Hydroxychlorothalonil ANM 2017 4-Hydroxydiphenylamine B19 1995 - 1997 5-Hydroxythiabendazole B28 1996 - 1997, 2003 - 2020 Abamectin 948 1994 - 1997, 1999, 2012 - 2020 Acephate 204 1991 - 2020 Acequinocyl AKS 2013 - 2015, 2020 Acetamiprid B80 2004 - 2020 Acetochlor 807 2001, 2003 - 2020 Acetochlor ethanesulfonic acid (ESA) ABN 2001 - 2013, 2017 Acetochlor oxanilic acid (OA) ABO 2001, 2003 - 2013 Acibenzolar S methyl B51 2003 - 2020 Acifluorfen 727 2003 - 2007, 2014 - 2017 Aclonifen D58 2017 - 2020 Acrinathrin A03 2012 - 2015, 2017 Afidopyropen F87 2020 Alachlor 227 1997 - 2020 Alachlor ethanesulfonic acid (ESA) ABP 2001 - 2013, 2017 Alachlor oxanilic acid (OA) ABQ 2001 - 2013 Aldicarb 167 1994 - 2020 Aldicarb sulfone 168 1994 - 2020 Aldicarb sulfoxide 169 1993 - 2020 Aldrin 001 1997 - 2020 Allethrin 002 1994, 1999