Analysis for Mutation Screening of RGSL1, RGS16 and RGS8 in Breast Cancer
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Related Malignant Phenotypes in the Nf1-Deficient MPNST
Published OnlineFirst February 19, 2013; DOI: 10.1158/1541-7786.MCR-12-0593 Molecular Cancer Genomics Research RAS/MEK–Independent Gene Expression Reveals BMP2- Related Malignant Phenotypes in the Nf1-Deficient MPNST Daochun Sun1, Ramsi Haddad2,3, Janice M. Kraniak2, Steven D. Horne1, and Michael A. Tainsky1,2 Abstract Malignant peripheral nerve sheath tumor (MPNST) is a type of soft tissue sarcoma that occurs in carriers of germline mutations in Nf1 gene as well as sporadically. Neurofibromin, encoded by the Nf1 gene, functions as a GTPase-activating protein (GAP) whose mutation leads to activation of wt-RAS and mitogen-activated protein kinase (MAPK) signaling in neurofibromatosis type I (NF1) patients' tumors. However, therapeutic targeting of RAS and MAPK have had limited success in this disease. In this study, we modulated NRAS, mitogen-activated protein/extracellular signal–regulated kinase (MEK)1/2, and neurofibromin levels in MPNST cells and determined gene expression changes to evaluate the regulation of signaling pathways in MPNST cells. Gene expression changes due to neurofibromin modulation but independent of NRAS and MEK1/2 regulation in MPNST cells indicated bone morphogenetic protein 2 (Bmp2) signaling as a key pathway. The BMP2-SMAD1/5/8 pathway was activated in NF1-associated MPNST cells and inhibition of BMP2 signaling by LDN-193189 or short hairpin RNA (shRNA) to BMP2 decreased the motility and invasion of NF1-associated MPNST cells. The pathway-specific gene changes provide a greater understanding of the complex role of neurofibromin in MPNST pathology and novel targets for drug discovery. Mol Cancer Res; 11(6); 616–27. -

Molecular Architecture of G O and the Structural Basis for RGS16
Molecular architecture of G␣o and the structural basis for RGS16-mediated deactivation Kevin C. Slep*†, Michele A. Kercher‡§, Thomas Wieland¶ʈ, Ching-Kang Chen¶, Melvin I. Simon¶**, and Paul B. Sigler‡§†† *Department of Biology, University of North Carolina, Chapel Hill, NC 27599; ‡Department of Molecular Biophysics and Biochemistry, and §Howard Hughes Medical Institute, Yale University, New Haven, CT 06511; and ¶Division of Biology, California Institute of Technology, Mail Code 147-75, Pasadena, CA 91125 Contributed by Melvin I. Simon, February 16, 2008 (sent for review December 18, 2007) Heterotrimeric G proteins relay extracellular cues from heptahelical (11). With the number of RGS proteins greatly exceeding the transmembrane receptors to downstream effector molecules. number of G␣ subunits, RGS proteins are likely to be finely tuned, Composed of an ␣ subunit with intrinsic GTPase activity and a ␥ titrated, and localized to regulate specific signaling pathways within heterodimer, the trimeric complex dissociates upon receptor- aG␣ subunit’s repertoire of effector targets. To mediate the mediated nucleotide exchange on the ␣ subunit, enabling each specificity and fidelity requisite for accurate signal transmission, the component to engage downstream effector targets for either G␣ subunit and its binding partners (GPCR, G␥, RGS, and activation or inhibition as dictated in a particular pathway. To effector) must have specific reciprocating molecular determinants mitigate excessive effector engagement and concomitant signal to minimize the convergence of independent signaling pathways. transmission, the G␣ subunit’s intrinsic activation timer (the rate of Thus, a comprehensive understanding of the molecular basis for G␣ GTP hydrolysis) is regulated spatially and temporally by a class of engagement with its activators, regulators, and effectors is critical GTPase accelerating proteins (GAPs) known as the regulator of G for elucidating specificity determinants. -

Identification of Transcriptional Mechanisms Downstream of Nf1 Gene Defeciency in Malignant Peripheral Nerve Sheath Tumors Daochun Sun Wayne State University
Wayne State University DigitalCommons@WayneState Wayne State University Dissertations 1-1-2012 Identification of transcriptional mechanisms downstream of nf1 gene defeciency in malignant peripheral nerve sheath tumors Daochun Sun Wayne State University, Follow this and additional works at: http://digitalcommons.wayne.edu/oa_dissertations Recommended Citation Sun, Daochun, "Identification of transcriptional mechanisms downstream of nf1 gene defeciency in malignant peripheral nerve sheath tumors" (2012). Wayne State University Dissertations. Paper 558. This Open Access Dissertation is brought to you for free and open access by DigitalCommons@WayneState. It has been accepted for inclusion in Wayne State University Dissertations by an authorized administrator of DigitalCommons@WayneState. IDENTIFICATION OF TRANSCRIPTIONAL MECHANISMS DOWNSTREAM OF NF1 GENE DEFECIENCY IN MALIGNANT PERIPHERAL NERVE SHEATH TUMORS by DAOCHUN SUN DISSERTATION Submitted to the Graduate School of Wayne State University, Detroit, Michigan in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY 2012 MAJOR: MOLECULAR BIOLOGY AND GENETICS Approved by: _______________________________________ Advisor Date _______________________________________ _______________________________________ _______________________________________ © COPYRIGHT BY DAOCHUN SUN 2012 All Rights Reserved DEDICATION This work is dedicated to my parents and my wife Ze Zheng for their continuous support and understanding during the years of my education. I could not achieve my goal without them. ii ACKNOWLEDGMENTS I would like to express tremendous appreciation to my mentor, Dr. Michael Tainsky. His guidance and encouragement throughout this project made this dissertation come true. I would also like to thank my committee members, Dr. Raymond Mattingly and Dr. John Reiners Jr. for their sustained attention to this project during the monthly NF1 group meetings and committee meetings, Dr. -

Supplementary Table 1
Supplementary Table 1. 492 genes are unique to 0 h post-heat timepoint. The name, p-value, fold change, location and family of each gene are indicated. Genes were filtered for an absolute value log2 ration 1.5 and a significance value of p ≤ 0.05. Symbol p-value Log Gene Name Location Family Ratio ABCA13 1.87E-02 3.292 ATP-binding cassette, sub-family unknown transporter A (ABC1), member 13 ABCB1 1.93E-02 −1.819 ATP-binding cassette, sub-family Plasma transporter B (MDR/TAP), member 1 Membrane ABCC3 2.83E-02 2.016 ATP-binding cassette, sub-family Plasma transporter C (CFTR/MRP), member 3 Membrane ABHD6 7.79E-03 −2.717 abhydrolase domain containing 6 Cytoplasm enzyme ACAT1 4.10E-02 3.009 acetyl-CoA acetyltransferase 1 Cytoplasm enzyme ACBD4 2.66E-03 1.722 acyl-CoA binding domain unknown other containing 4 ACSL5 1.86E-02 −2.876 acyl-CoA synthetase long-chain Cytoplasm enzyme family member 5 ADAM23 3.33E-02 −3.008 ADAM metallopeptidase domain Plasma peptidase 23 Membrane ADAM29 5.58E-03 3.463 ADAM metallopeptidase domain Plasma peptidase 29 Membrane ADAMTS17 2.67E-04 3.051 ADAM metallopeptidase with Extracellular other thrombospondin type 1 motif, 17 Space ADCYAP1R1 1.20E-02 1.848 adenylate cyclase activating Plasma G-protein polypeptide 1 (pituitary) receptor Membrane coupled type I receptor ADH6 (includes 4.02E-02 −1.845 alcohol dehydrogenase 6 (class Cytoplasm enzyme EG:130) V) AHSA2 1.54E-04 −1.6 AHA1, activator of heat shock unknown other 90kDa protein ATPase homolog 2 (yeast) AK5 3.32E-02 1.658 adenylate kinase 5 Cytoplasm kinase AK7 -

Dissertation Kollipara.Pdf
Characterizing protein processing in the Endoplasmic Reticulum using quantitative proteomics: the pathogenesis of the Marinesco-Sjörgren Syndrome Zur Erlangung des akademischen Grades eines Dr. rer. nat von der Fakultät Bio- und Chemieingenieurwesen der Technischen Universität Dortmund genehmigte Dissertation vorgelegt von MSc. Laxmikanth Kollipara aus Hyderabad, India Tag der mündlichen Prüfung: 20.12.2016 1. Gutachter: Prof. Dr. Albert Sickmann 2. Gutachter: Prof. Dr. Oliver Kayser Dortmund 2016 1. Prüfer: Prof. Dr. Markus Nett Abstract Abstract In this work, characterization of Marinesco-Sjögren Syndrome (MSS) was performed for the first time on the proteome level using mass spectrometry (MS)-based quantitative proteomics strategies. MSS is a neuromuscular and neurodegenerative disorder and it is caused due to the mutational inactivation of SIL1 protein, which results in malfunctioning of protein folding machinery mediated by the chaperone BiP that can lead to the ER stress-induced cell death via apoptotic signaling. The major goals were (i) to understand the rescue mechanisms in SIL1-deficient non-vulnerable tissues from human and (ii) to verify the cellular perturbations caused due to the loss of functional SIL1 in woozy mouse (i.e. mouse model of MSS). To achieve these aims, samples derived from five different MSS cases and two different tissues from woozy along with their respective healthy controls were studied. For this, comparative LC-MS proteomics approaches such as chemical labeling (i.e. iTRAQ) and label- free quantification (precursor ion intensity based and NSAF) were employed. During which, sample preparation workflows were optimized that enabled to process clinical samples related to MSS that included primary cell lines and mammalian tissues for the subsequent quantitative LC-MS analyses. -

Egfr Activates a Taz-Driven Oncogenic Program in Glioblastoma
EGFR ACTIVATES A TAZ-DRIVEN ONCOGENIC PROGRAM IN GLIOBLASTOMA by Minling Gao A thesis submitted to Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland March 2020 ©2020 Minling Gao All rights reserved Abstract Hyperactivated EGFR signaling is associated with about 45% of Glioblastoma (GBM), the most aggressive and lethal primary brain tumor in humans. However, the oncogenic transcriptional events driven by EGFR are still incompletely understood. We studied the role of the transcription factor TAZ to better understand master transcriptional regulators in mediating the EGFR signaling pathway in GBM. The transcriptional coactivator with PDZ- binding motif (TAZ) and its paralog gene, the Yes-associated protein (YAP) are two transcriptional co-activators that play important roles in multiple cancer types and are regulated in a context-dependent manner by various upstream signaling pathways, e.g. the Hippo, WNT and GPCR signaling. In GBM cells, TAZ functions as an oncogene that drives mesenchymal transition and radioresistance. This thesis intends to broaden our understanding of EGFR signaling and TAZ regulation in GBM. In patient-derived GBM cell models, EGF induced TAZ and its known gene targets through EGFR and downstream tyrosine kinases (ERK1/2 and STAT3). In GBM cells with EGFRvIII, an EGF-independent and constitutively active mutation, TAZ showed EGF- independent hyperactivation when compared to EGFRvIII-negative cells. These results revealed a novel EGFR-TAZ signaling axis in GBM cells. The second contribution of this thesis is that we performed next-generation sequencing to establish the first genome-wide map of EGF-induced TAZ target genes. -
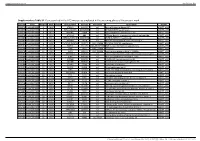
Supplementary Table S1. Genes Printed in the HC5 Microarray Employed in the Screening Phase of the Present Work
Supplementary material Ann Rheum Dis Supplementary Table S1. Genes printed in the HC5 microarray employed in the screening phase of the present work. CloneID Plate Position Well Length GeneSymbol GeneID Accession Description Vector 692672 HsxXG013989 2 B01 STK32A 202374 null serine/threonine kinase 32A pANT7_cGST 692675 HsxXG013989 3 C01 RPS10-NUDT3 100529239 null RPS10-NUDT3 readthrough pANT7_cGST 692678 HsxXG013989 4 D01 SPATA6L 55064 null spermatogenesis associated 6-like pANT7_cGST 692679 HsxXG013989 5 E01 ATP1A4 480 null ATPase, Na+/K+ transporting, alpha 4 polypeptide pANT7_cGST 692689 HsxXG013989 6 F01 ZNF816-ZNF321P 100529240 null ZNF816-ZNF321P readthrough pANT7_cGST 692691 HsxXG013989 7 G01 NKAIN1 79570 null Na+/K+ transporting ATPase interacting 1 pANT7_cGST 693155 HsxXG013989 8 H01 TNFSF12-TNFSF13 407977 NM_172089 TNFSF12-TNFSF13 readthrough pANT7_cGST 693161 HsxXG013989 9 A02 RAB12 201475 NM_001025300 RAB12, member RAS oncogene family pANT7_cGST 693169 HsxXG013989 10 B02 SYN1 6853 NM_133499 synapsin I pANT7_cGST 693176 HsxXG013989 11 C02 GJD3 125111 NM_152219 gap junction protein, delta 3, 31.9kDa pANT7_cGST 693181 HsxXG013989 12 D02 CHCHD10 400916 null coiled-coil-helix-coiled-coil-helix domain containing 10 pANT7_cGST 693184 HsxXG013989 13 E02 IDNK 414328 null idnK, gluconokinase homolog (E. coli) pANT7_cGST 693187 HsxXG013989 14 F02 LYPD6B 130576 null LY6/PLAUR domain containing 6B pANT7_cGST 693189 HsxXG013989 15 G02 C8orf86 389649 null chromosome 8 open reading frame 86 pANT7_cGST 693194 HsxXG013989 16 H02 CENPQ 55166 -

The Human Gene Connectome As a Map of Short Cuts for Morbid Allele Discovery
The human gene connectome as a map of short cuts for morbid allele discovery Yuval Itana,1, Shen-Ying Zhanga,b, Guillaume Vogta,b, Avinash Abhyankara, Melina Hermana, Patrick Nitschkec, Dror Friedd, Lluis Quintana-Murcie, Laurent Abela,b, and Jean-Laurent Casanovaa,b,f aSt. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, NY 10065; bLaboratory of Human Genetics of Infectious Diseases, Necker Branch, Paris Descartes University, Institut National de la Santé et de la Recherche Médicale U980, Necker Medical School, 75015 Paris, France; cPlateforme Bioinformatique, Université Paris Descartes, 75116 Paris, France; dDepartment of Computer Science, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel; eUnit of Human Evolutionary Genetics, Centre National de la Recherche Scientifique, Unité de Recherche Associée 3012, Institut Pasteur, F-75015 Paris, France; and fPediatric Immunology-Hematology Unit, Necker Hospital for Sick Children, 75015 Paris, France Edited* by Bruce Beutler, University of Texas Southwestern Medical Center, Dallas, TX, and approved February 15, 2013 (received for review October 19, 2012) High-throughput genomic data reveal thousands of gene variants to detect a single mutated gene, with the other polymorphic genes per patient, and it is often difficult to determine which of these being of less interest. This goes some way to explaining why, variants underlies disease in a given individual. However, at the despite the abundance of NGS data, the discovery of disease- population level, there may be some degree of phenotypic homo- causing alleles from such data remains somewhat limited. geneity, with alterations of specific physiological pathways under- We developed the human gene connectome (HGC) to over- come this problem. -

NIH Public Access Author Manuscript Schizophr Res
NIH Public Access Author Manuscript Schizophr Res. Author manuscript; available in PMC 2009 April 1. NIH-PA Author ManuscriptPublished NIH-PA Author Manuscript in final edited NIH-PA Author Manuscript form as: Schizophr Res. 2008 April ; 101(1-3): 67±75. ASSOCIATION OF RGS2 AND RGS5 VARIANTS WITH SCHIZOPHRENIA SYMPTOM SEVERITY Daniel B. Campbell1, Leslie A. Lange2, Tara Skelly2, Jeffrey Lieberman3, Pat Levitt1,4, and Patrick F. Sullivan2,5 1Department of Pharmacology, Vanderbilt University, Nashville, TN 37232 USA 2Department of Genetics, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599 USA 3Department of Psychiatry, Columbia University, New York, NY 10032 USA 4Vanderbilt Kennedy Center for Research on Human Development, Vanderbilt University, Nashville, TN 37203 USA 5Department of Medical Epidemiology & Biostatistics, Karolinska Institutet, SE-171 77 Stockholm, Sweden Abstract Background—Several lines of evidence indicate that Regulator of G Protein Signaling 4 (RGS4) contributes to schizophrenia vulnerability. RGS4 is one of a family of molecules that modulate signaling via G-protein coupled receptors. Five genes encoding members of this family (RGS2, RGS4, RGS5, RGS8 and RGS16) map to chromosome 1q23.3-1q31. Due to overlapping cellular functions and chromosomal proximity, we hypothesized that multiple RGS genes may contribute to schizophrenia severity and treatment responsiveness. Methods—Subjects were 750 individuals with schizophrenia who participated in the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE). Inferred ancestries were: 221 (30%) ‘Africa only’, 422 (56%) ‘Europe only’ and 107 (14%) ‘Other’. Fifty-nine single nucleotide polymorphisms (SNPs) in or near the RGS5, RGS16, RGS8 and RGS2 genes were genotyped. Multiple linear regression was used to analyze association of markers with Positive and Negative Symptoms Scale (PANSS) total scores at baseline and throughout antipsychotic treatment. -

Regulators of G-Protein Signaling (RGS) Proteins: Region-Specific Expression of Nine Subtypes in Rat Brain
The Journal of Neuroscience, October 15, 1997, 17(20):8024–8037 Regulators of G-Protein Signaling (RGS) Proteins: Region-Specific Expression of Nine Subtypes in Rat Brain Stephen J. Gold,1,2 YanG.Ni,1,2 Henrik G. Dohlman,3 and Eric J. Nestler1,2,3 1Laboratory of Molecular Psychiatry and Departments of 2Psychiatry and 3Pharmacology, Yale University School of Medicine, New Haven, Connecticut 06508 The recently discovered regulators of G-protein signaling (RGS) richment in striatal regions. RGS10 mRNA is densely expressed proteins potently modulate the functioning of heterotrimeric in dentate gyrus granule cells, superficial layers of neocortex, G-proteins by stimulating the GTPase activity of G-protein a and dorsal raphe. To assess whether the expression of RGS subunits. The mRNAs for numerous subtypes of putative RGS mRNAs can be regulated, we examined the effect of an acute proteins have been identified in mammalian tissues, but little is seizure on levels of RGS7, RGS8, and RGS10 mRNAs in hip- known about their expression in brain. We performed a system- pocampus. Of the three subtypes, changes in RGS10 levels atic survey of the localization of mRNAs encoding nine of these were most pronounced, decreasing by ;40% in a time- RGSs, RGS3–RGS11, in brain by in situ hybridization. Striking dependent manner in response to a single seizure. These re- region-specific patterns of expression were observed. Five sults, which document highly specific patterns of RGS mRNA subtypes, RGS4, RGS7, RGS8, RGS9, and RGS10 mRNAs, are expression in brain and their ability to be regulated in a dynamic densely expressed in brain, whereas the other subtypes (RGS3, manner, support the view that RGS proteins may play an im- RGS5, RGS6, and RGS11) are expressed at lower density and portant role in determining the intensity and specificity of sig- in more restricted regions. -

The Role of the Chaperone CCT in Assembling Cell Signaling Complexes
Brigham Young University BYU ScholarsArchive Theses and Dissertations 2020-07-21 The Role of the Chaperone CCT in Assembling Cell Signaling Complexes Nicole C. Tensmeyer Brigham Young University Follow this and additional works at: https://scholarsarchive.byu.edu/etd Part of the Physical Sciences and Mathematics Commons BYU ScholarsArchive Citation Tensmeyer, Nicole C., "The Role of the Chaperone CCT in Assembling Cell Signaling Complexes" (2020). Theses and Dissertations. 9192. https://scholarsarchive.byu.edu/etd/9192 This Dissertation is brought to you for free and open access by BYU ScholarsArchive. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of BYU ScholarsArchive. For more information, please contact [email protected]. The Role of the Molecular Chaperone CCT in Assembling Cell Signaling Complexes Nicole C. Tensmeyer A dissertation submitted to the faculty of Brigham Young University in partial fulfillment of the requirements for the degree of Doctor of Philosophy Barry M. Willardson, Chair Josh L. Andersen John C. Price Pam Van Ry Department of Chemistry and Biochemistry Brigham Young University Copyright © 2020 Nicole C. Tensmeyer All Rights Reserved ABSTRACT The Role of the Molecular Chaperone CCT in the Assembly of Signaling Complexes Nicole C. Tensmeyer Department of Chemistry and Biochemistry, BYU Doctor of Philosophy In order to function, proteins must be folded into their native shape. While this can sometimes occur spontaneously, the process can be hindered by thermodynamic barriers, trapped intermediates, and aggregation prone hydrophobic interactions. Molecular chaperones are proteins that help client proteins or substrates overcome these barriers so that they can be folded properly.