Dissertation Kollipara.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Table 2. Significant
Table 2. Significant (Q < 0.05 and |d | > 0.5) transcripts from the meta-analysis Gene Chr Mb Gene Name Affy ProbeSet cDNA_IDs d HAP/LAP d HAP/LAP d d IS Average d Ztest P values Q-value Symbol ID (study #5) 1 2 STS B2m 2 122 beta-2 microglobulin 1452428_a_at AI848245 1.75334941 4 3.2 4 3.2316485 1.07398E-09 5.69E-08 Man2b1 8 84.4 mannosidase 2, alpha B1 1416340_a_at H4049B01 3.75722111 3.87309653 2.1 1.6 2.84852656 5.32443E-07 1.58E-05 1110032A03Rik 9 50.9 RIKEN cDNA 1110032A03 gene 1417211_a_at H4035E05 4 1.66015788 4 1.7 2.82772795 2.94266E-05 0.000527 NA 9 48.5 --- 1456111_at 3.43701477 1.85785922 4 2 2.8237185 9.97969E-08 3.48E-06 Scn4b 9 45.3 Sodium channel, type IV, beta 1434008_at AI844796 3.79536664 1.63774235 3.3 2.3 2.75319499 1.48057E-08 6.21E-07 polypeptide Gadd45gip1 8 84.1 RIKEN cDNA 2310040G17 gene 1417619_at 4 3.38875643 1.4 2 2.69163229 8.84279E-06 0.0001904 BC056474 15 12.1 Mus musculus cDNA clone 1424117_at H3030A06 3.95752801 2.42838452 1.9 2.2 2.62132809 1.3344E-08 5.66E-07 MGC:67360 IMAGE:6823629, complete cds NA 4 153 guanine nucleotide binding protein, 1454696_at -3.46081884 -4 -1.3 -1.6 -2.6026947 8.58458E-05 0.0012617 beta 1 Gnb1 4 153 guanine nucleotide binding protein, 1417432_a_at H3094D02 -3.13334396 -4 -1.6 -1.7 -2.5946297 1.04542E-05 0.0002202 beta 1 Gadd45gip1 8 84.1 RAD23a homolog (S. -

An Update on the Metabolic Roles of Carbonic Anhydrases in the Model Alga Chlamydomonas Reinhardtii
H OH metabolites OH Review An Update on the Metabolic Roles of Carbonic Anhydrases in the Model Alga Chlamydomonas reinhardtii Ashok Aspatwar 1,* ID , Susanna Haapanen 1 and Seppo Parkkila 1,2 1 Faculty of Medicine and Life Sciences, University of Tampere, FI-33014 Tampere, Finland; [email protected].fi (S.H.); [email protected].fi (S.P.) 2 Fimlab, Ltd., and Tampere University Hospital, FI-33520 Tampere, Finland * Correspondence: [email protected].fi; Tel.: +358-46-596-2117 Received: 11 January 2018; Accepted: 10 March 2018; Published: 13 March 2018 Abstract: Carbonic anhydrases (CAs) are metalloenzymes that are omnipresent in nature. − + CAs catalyze the basic reaction of the reversible hydration of CO2 to HCO3 and H in all living organisms. Photosynthetic organisms contain six evolutionarily different classes of CAs, which are namely: α-CAs, β-CAs, γ-CAs, δ-CAs, ζ-CAs, and θ-CAs. Many of the photosynthetic organisms contain multiple isoforms of each CA family. The model alga Chlamydomonas reinhardtii contains 15 CAs belonging to three different CA gene families. Of these 15 CAs, three belong to the α-CA gene family; nine belong to the β-CA gene family; and three belong to the γ-CA gene family. The multiple copies of the CAs in each gene family may be due to gene duplications within the particular CA gene family. The CAs of Chlamydomonas reinhardtii are localized in different subcellular compartments of this unicellular alga. The presence of a large number of CAs and their diverse subcellular localization within a single cell suggests the importance of these enzymes in the metabolic and biochemical roles they perform in this unicellular alga. -

Supplementary Table 1
Supplementary Table 1. 492 genes are unique to 0 h post-heat timepoint. The name, p-value, fold change, location and family of each gene are indicated. Genes were filtered for an absolute value log2 ration 1.5 and a significance value of p ≤ 0.05. Symbol p-value Log Gene Name Location Family Ratio ABCA13 1.87E-02 3.292 ATP-binding cassette, sub-family unknown transporter A (ABC1), member 13 ABCB1 1.93E-02 −1.819 ATP-binding cassette, sub-family Plasma transporter B (MDR/TAP), member 1 Membrane ABCC3 2.83E-02 2.016 ATP-binding cassette, sub-family Plasma transporter C (CFTR/MRP), member 3 Membrane ABHD6 7.79E-03 −2.717 abhydrolase domain containing 6 Cytoplasm enzyme ACAT1 4.10E-02 3.009 acetyl-CoA acetyltransferase 1 Cytoplasm enzyme ACBD4 2.66E-03 1.722 acyl-CoA binding domain unknown other containing 4 ACSL5 1.86E-02 −2.876 acyl-CoA synthetase long-chain Cytoplasm enzyme family member 5 ADAM23 3.33E-02 −3.008 ADAM metallopeptidase domain Plasma peptidase 23 Membrane ADAM29 5.58E-03 3.463 ADAM metallopeptidase domain Plasma peptidase 29 Membrane ADAMTS17 2.67E-04 3.051 ADAM metallopeptidase with Extracellular other thrombospondin type 1 motif, 17 Space ADCYAP1R1 1.20E-02 1.848 adenylate cyclase activating Plasma G-protein polypeptide 1 (pituitary) receptor Membrane coupled type I receptor ADH6 (includes 4.02E-02 −1.845 alcohol dehydrogenase 6 (class Cytoplasm enzyme EG:130) V) AHSA2 1.54E-04 −1.6 AHA1, activator of heat shock unknown other 90kDa protein ATPase homolog 2 (yeast) AK5 3.32E-02 1.658 adenylate kinase 5 Cytoplasm kinase AK7 -
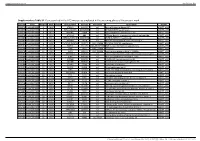
Supplementary Table S1. Genes Printed in the HC5 Microarray Employed in the Screening Phase of the Present Work
Supplementary material Ann Rheum Dis Supplementary Table S1. Genes printed in the HC5 microarray employed in the screening phase of the present work. CloneID Plate Position Well Length GeneSymbol GeneID Accession Description Vector 692672 HsxXG013989 2 B01 STK32A 202374 null serine/threonine kinase 32A pANT7_cGST 692675 HsxXG013989 3 C01 RPS10-NUDT3 100529239 null RPS10-NUDT3 readthrough pANT7_cGST 692678 HsxXG013989 4 D01 SPATA6L 55064 null spermatogenesis associated 6-like pANT7_cGST 692679 HsxXG013989 5 E01 ATP1A4 480 null ATPase, Na+/K+ transporting, alpha 4 polypeptide pANT7_cGST 692689 HsxXG013989 6 F01 ZNF816-ZNF321P 100529240 null ZNF816-ZNF321P readthrough pANT7_cGST 692691 HsxXG013989 7 G01 NKAIN1 79570 null Na+/K+ transporting ATPase interacting 1 pANT7_cGST 693155 HsxXG013989 8 H01 TNFSF12-TNFSF13 407977 NM_172089 TNFSF12-TNFSF13 readthrough pANT7_cGST 693161 HsxXG013989 9 A02 RAB12 201475 NM_001025300 RAB12, member RAS oncogene family pANT7_cGST 693169 HsxXG013989 10 B02 SYN1 6853 NM_133499 synapsin I pANT7_cGST 693176 HsxXG013989 11 C02 GJD3 125111 NM_152219 gap junction protein, delta 3, 31.9kDa pANT7_cGST 693181 HsxXG013989 12 D02 CHCHD10 400916 null coiled-coil-helix-coiled-coil-helix domain containing 10 pANT7_cGST 693184 HsxXG013989 13 E02 IDNK 414328 null idnK, gluconokinase homolog (E. coli) pANT7_cGST 693187 HsxXG013989 14 F02 LYPD6B 130576 null LY6/PLAUR domain containing 6B pANT7_cGST 693189 HsxXG013989 15 G02 C8orf86 389649 null chromosome 8 open reading frame 86 pANT7_cGST 693194 HsxXG013989 16 H02 CENPQ 55166 -

NIH Public Access Author Manuscript Schizophr Res
NIH Public Access Author Manuscript Schizophr Res. Author manuscript; available in PMC 2009 April 1. NIH-PA Author ManuscriptPublished NIH-PA Author Manuscript in final edited NIH-PA Author Manuscript form as: Schizophr Res. 2008 April ; 101(1-3): 67±75. ASSOCIATION OF RGS2 AND RGS5 VARIANTS WITH SCHIZOPHRENIA SYMPTOM SEVERITY Daniel B. Campbell1, Leslie A. Lange2, Tara Skelly2, Jeffrey Lieberman3, Pat Levitt1,4, and Patrick F. Sullivan2,5 1Department of Pharmacology, Vanderbilt University, Nashville, TN 37232 USA 2Department of Genetics, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599 USA 3Department of Psychiatry, Columbia University, New York, NY 10032 USA 4Vanderbilt Kennedy Center for Research on Human Development, Vanderbilt University, Nashville, TN 37203 USA 5Department of Medical Epidemiology & Biostatistics, Karolinska Institutet, SE-171 77 Stockholm, Sweden Abstract Background—Several lines of evidence indicate that Regulator of G Protein Signaling 4 (RGS4) contributes to schizophrenia vulnerability. RGS4 is one of a family of molecules that modulate signaling via G-protein coupled receptors. Five genes encoding members of this family (RGS2, RGS4, RGS5, RGS8 and RGS16) map to chromosome 1q23.3-1q31. Due to overlapping cellular functions and chromosomal proximity, we hypothesized that multiple RGS genes may contribute to schizophrenia severity and treatment responsiveness. Methods—Subjects were 750 individuals with schizophrenia who participated in the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE). Inferred ancestries were: 221 (30%) ‘Africa only’, 422 (56%) ‘Europe only’ and 107 (14%) ‘Other’. Fifty-nine single nucleotide polymorphisms (SNPs) in or near the RGS5, RGS16, RGS8 and RGS2 genes were genotyped. Multiple linear regression was used to analyze association of markers with Positive and Negative Symptoms Scale (PANSS) total scores at baseline and throughout antipsychotic treatment. -

Regulators of G-Protein Signaling (RGS) Proteins: Region-Specific Expression of Nine Subtypes in Rat Brain
The Journal of Neuroscience, October 15, 1997, 17(20):8024–8037 Regulators of G-Protein Signaling (RGS) Proteins: Region-Specific Expression of Nine Subtypes in Rat Brain Stephen J. Gold,1,2 YanG.Ni,1,2 Henrik G. Dohlman,3 and Eric J. Nestler1,2,3 1Laboratory of Molecular Psychiatry and Departments of 2Psychiatry and 3Pharmacology, Yale University School of Medicine, New Haven, Connecticut 06508 The recently discovered regulators of G-protein signaling (RGS) richment in striatal regions. RGS10 mRNA is densely expressed proteins potently modulate the functioning of heterotrimeric in dentate gyrus granule cells, superficial layers of neocortex, G-proteins by stimulating the GTPase activity of G-protein a and dorsal raphe. To assess whether the expression of RGS subunits. The mRNAs for numerous subtypes of putative RGS mRNAs can be regulated, we examined the effect of an acute proteins have been identified in mammalian tissues, but little is seizure on levels of RGS7, RGS8, and RGS10 mRNAs in hip- known about their expression in brain. We performed a system- pocampus. Of the three subtypes, changes in RGS10 levels atic survey of the localization of mRNAs encoding nine of these were most pronounced, decreasing by ;40% in a time- RGSs, RGS3–RGS11, in brain by in situ hybridization. Striking dependent manner in response to a single seizure. These re- region-specific patterns of expression were observed. Five sults, which document highly specific patterns of RGS mRNA subtypes, RGS4, RGS7, RGS8, RGS9, and RGS10 mRNAs, are expression in brain and their ability to be regulated in a dynamic densely expressed in brain, whereas the other subtypes (RGS3, manner, support the view that RGS proteins may play an im- RGS5, RGS6, and RGS11) are expressed at lower density and portant role in determining the intensity and specificity of sig- in more restricted regions. -

S1 of S77 Supplementary Materials: Discovery of a New Class of Cathepsin Kinhibitors in Rhizoma Drynariaeas Potential Candidates for the Treatment of Osteoporosis
Int. J. Mol. Sci.2016, 17, 2116; doi:10.3390/ijms17122116 S1 of S77 Supplementary Materials: Discovery of a New Class of Cathepsin KInhibitors in Rhizoma Drynariaeas Potential Candidates for the Treatment of Osteoporosis Zuo-Cheng Qiu, Xiao-Li Dong, Yi Dai, Gao-Keng Xiao, Xin-Luan Wang, Ka-Chun Wong, Man-Sau Wong and Xin-Sheng Yao Table S1. Compounds identified from Drynariae rhizome (DR). No. Compound Name Chemical Structure 1 Naringin 5,7,3′,5′-Tetrahydroxy-flavanone 2 7-O-neohesperidoside 3 Narigenin-7-O-β-D-glucoside 5,7,3′,5′-Tetrahydroxy-flavanone 4 7-O-β-D-glucopyranoside 5 Naringenin 6 5,7,3′,5′-Tetrahydroxyflavanone 7 Kushennol F 8 Sophoraflavanone G 9 Kurarinone Int. J. Mol. Sci.2016, 17, 2116; doi:10.3390/ijms17122116 S2 of S77 Table S1. Cont. No. Compound Name Chemical Structure 10 Leachianone A 11 Luteolin-7-O-neohesperidoside 12 Luteolin-5-O-neohesperidoside 13 Kaempferol-7-O-α-L-arabinofuranoside 14 8-Prenylapigenin 15 Apigenine 16 Kaempferol-3-O-α-L-rhamnopyranoside OH HO O 17 Astragalin O OH OH O O OH OH OH 18 3-O-β-D-Glucopyranoside-7-O-α-L-arabinofuranoside OH HO O HO O O 19 5,7-Dihydroxychromone-7-O-β-D-glucopyranoside OH OH O Int. J. Mol. Sci.2016, 17, 2116; doi:10.3390/ijms17122116 S3 of S77 Table S1. Cont. No. Compound Name Chemical Structure 20 5,7-Dihydroxychromone-7-O-neohesperidoside Kaempferol 21 3-O-β-D-glucopyranoside-7-O-β-D-glucopyranoside 22 Xanthohumol OH HO O 23 Epicatechin OH OH OH 24 (E)-4-O-β-D-Glucopyranosyl caffeic acid 25 β-D-Glucopyranosyl sinapoic acid 26 4-O-β-D-Glucopyranosyl ferulic acid 27 Trans-caffeic acid 28 4-O-β-D-Glucopyranosyl coumaric acid 29 Dihydrocaffeic acid methyl ester 30 Dihydrocaffeic acid 31 3,4-Dihydroxyl benzoic acid 32 4-O-D-Glucosyl vanillic acid Int. -

The Role of the Chaperone CCT in Assembling Cell Signaling Complexes
Brigham Young University BYU ScholarsArchive Theses and Dissertations 2020-07-21 The Role of the Chaperone CCT in Assembling Cell Signaling Complexes Nicole C. Tensmeyer Brigham Young University Follow this and additional works at: https://scholarsarchive.byu.edu/etd Part of the Physical Sciences and Mathematics Commons BYU ScholarsArchive Citation Tensmeyer, Nicole C., "The Role of the Chaperone CCT in Assembling Cell Signaling Complexes" (2020). Theses and Dissertations. 9192. https://scholarsarchive.byu.edu/etd/9192 This Dissertation is brought to you for free and open access by BYU ScholarsArchive. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of BYU ScholarsArchive. For more information, please contact [email protected]. The Role of the Molecular Chaperone CCT in Assembling Cell Signaling Complexes Nicole C. Tensmeyer A dissertation submitted to the faculty of Brigham Young University in partial fulfillment of the requirements for the degree of Doctor of Philosophy Barry M. Willardson, Chair Josh L. Andersen John C. Price Pam Van Ry Department of Chemistry and Biochemistry Brigham Young University Copyright © 2020 Nicole C. Tensmeyer All Rights Reserved ABSTRACT The Role of the Molecular Chaperone CCT in the Assembly of Signaling Complexes Nicole C. Tensmeyer Department of Chemistry and Biochemistry, BYU Doctor of Philosophy In order to function, proteins must be folded into their native shape. While this can sometimes occur spontaneously, the process can be hindered by thermodynamic barriers, trapped intermediates, and aggregation prone hydrophobic interactions. Molecular chaperones are proteins that help client proteins or substrates overcome these barriers so that they can be folded properly. -

Downregulation of Carnitine Acyl-Carnitine Translocase by Mirnas
Page 1 of 288 Diabetes 1 Downregulation of Carnitine acyl-carnitine translocase by miRNAs 132 and 212 amplifies glucose-stimulated insulin secretion Mufaddal S. Soni1, Mary E. Rabaglia1, Sushant Bhatnagar1, Jin Shang2, Olga Ilkayeva3, Randall Mynatt4, Yun-Ping Zhou2, Eric E. Schadt6, Nancy A.Thornberry2, Deborah M. Muoio5, Mark P. Keller1 and Alan D. Attie1 From the 1Department of Biochemistry, University of Wisconsin, Madison, Wisconsin; 2Department of Metabolic Disorders-Diabetes, Merck Research Laboratories, Rahway, New Jersey; 3Sarah W. Stedman Nutrition and Metabolism Center, Duke Institute of Molecular Physiology, 5Departments of Medicine and Pharmacology and Cancer Biology, Durham, North Carolina. 4Pennington Biomedical Research Center, Louisiana State University system, Baton Rouge, Louisiana; 6Institute for Genomics and Multiscale Biology, Mount Sinai School of Medicine, New York, New York. Corresponding author Alan D. Attie, 543A Biochemistry Addition, 433 Babcock Drive, Department of Biochemistry, University of Wisconsin-Madison, Madison, Wisconsin, (608) 262-1372 (Ph), (608) 263-9608 (fax), [email protected]. Running Title: Fatty acyl-carnitines enhance insulin secretion Abstract word count: 163 Main text Word count: 3960 Number of tables: 0 Number of figures: 5 Diabetes Publish Ahead of Print, published online June 26, 2014 Diabetes Page 2 of 288 2 ABSTRACT We previously demonstrated that micro-RNAs 132 and 212 are differentially upregulated in response to obesity in two mouse strains that differ in their susceptibility to obesity-induced diabetes. Here we show the overexpression of micro-RNAs 132 and 212 enhances insulin secretion (IS) in response to glucose and other secretagogues including non-fuel stimuli. We determined that carnitine acyl-carnitine translocase (CACT, Slc25a20) is a direct target of these miRNAs. -

Alternative Splicing of RGS8 Gene Determines Inhibitory Function of Receptor Type-Specific Gq Signaling
Alternative splicing of RGS8 gene determines inhibitory function of receptor type-specific Gq signaling Osamu Saitoh*†, Yoshimichi Murata‡, Megumi Odagiri*, Masayuki Itoh*§, Hiroshi Itoh¶, Takumi Misaka‡ʈ, and Yoshihiro Kubo‡ *Department of Molecular Cell Signaling, Tokyo Metropolitan Institute for Neuroscience, 2-6 Musashidai, Fuchu-shi, Tokyo 183-8526, Japan; ‡Department of Physiology and Cell Biology, Tokyo Medical and Dental University, Graduate School and Faculty of Medicine, Yushima, Bunkyo-ku, Tokyo 113-8519, Japan; §Department of Biomolecular Science, Faculty of Science, Toho University, Miyama, Funabashi, Chiba 274-8510, Japan; ¶Graduate School of Biological Sciences, Nara Institute of Science and Technology, 8916-5 Takayama, Ikoma, Nara 630-0101, Japan; and ʈDepartment of Applied Biological Chemistry, Graduate School of Agricultural and Life Sciences, University of Tokyo, Yayoi, Bunkyo-ku, Tokyo 113-8657, Japan Edited by Lily Y. Jan, University of California School of Medicine, San Francisco, CA, and approved May 23, 2002 (received for review February 12, 2002) The regulators of G protein signaling (RGS) proteins modulate signaling. There is significant sequence conservation in the heterotrimeric G protein signaling. RGS8 is a brain-specific RGS N-terminal region of RGS4 and RGS8 (6). protein of 180 aa. Here we identified a short isoform of RGS8, It was recently reported that RGS3 is expressed as two RGS8S, that arises by alternative splicing. RGS8S cDNA encodes a isoforms transcribed from alternate promoter sites within the N terminus of 7 aa instead of amino acids 1–9 of RGS8 and 10–180 RGS3 gene and that the RGS3T form lacks a large portion of the of RGS8. -

Supplementary Table 2: Infection-Sensitive Genes
Supplementary Table 2: Infection-Sensitive Genes Supplementary Table 2: Upregulated with Adenoviral Infection (pages 1 - 7)- genes that were significant by ANOVA as well as significantly increased in control compared to both 'infected control' (Ad-LacZ) and 'calcineurin infected' (Ad-aCaN). Downregulated with Adenoviral Infection (pages 7 - 13) as above except for direction of change. Columns: Probe Set- Affymetrix probe set identifier for RG-U34A microarray, Symbol and Title- annotated information for above probe set (annotation downloaded June, 2004), ANOVA- p-value for 1-way Analysis of Variance, Uninfected, Ad-LacZ and Ad- aCaN- mean ± SEM expression for uninfected control, adenovirus mediated LacZ treated control, and adenovirus mediated calcineurin treated cultures respectively. Upregulated with Adenoviral Infection Probe Set Symbol Title ANOVA Uninfected Ad-LacZ Ad-aCaN Z48444cds_at Adam10 a disintegrin and metalloprotease domain 10 0.00001 837 ± 31 1107 ± 24 1028 ± 29 L26986_at Adcy8 adenylyl cyclase 8 0.00028 146 ± 12 252 ± 21 272 ± 21 U94340_at Adprt ADP-ribosyltransferase 1 0.00695 2096 ± 100 2783 ± 177 2586 ± 118 U01914_at Akap8 A kinase anchor protein 8 0.00080 993 ± 44 1194 ± 65 1316 ± 44 AB008538_at Alcam activated leukocyte cell adhesion molecule 0.01012 2775 ± 136 3580 ± 216 3429 ± 155 M34176_s_at Ap2b1 adaptor-related protein complex 2, beta 1 subunit 0.01389 2408 ± 199 2947 ± 143 3071 ± 116 D44495_s_at Apex1 apurinic/apyrimidinic endonuclease 1 0.00089 4959 ± 185 5816 ± 202 6057 ± 158 U16245_at Aqp5 aquaporin 5 0.02710 -

Roles for Regulator of G Protein Signaling Proteins in Synaptic Signaling and Plasticity
1521-0111/89/2/273–286$25.00 http://dx.doi.org/10.1124/mol.115.102210 MOLECULAR PHARMACOLOGY Mol Pharmacol 89:273–286, February 2016 Copyright ª 2016 by The American Society for Pharmacology and Experimental Therapeutics MINIREVIEW Roles for Regulator of G Protein Signaling Proteins in Synaptic Signaling and Plasticity Kyle J. Gerber, Katherine E. Squires, and John R. Hepler Programs in Molecular and Systems Pharmacology (K.J.G., K.E.S., J.R.H.) and Neuroscience (J.R.H.), Department of Downloaded from Pharmacology (K.J.G., K.E.S., J.R.H.), Emory University School of Medicine, Atlanta, Georgia Received October 26, 2015; accepted December 10, 2015 ABSTRACT The regulator of G protein signaling (RGS) family of proteins synaptic transmission, and synaptic plasticity, which are neces- molpharm.aspetjournals.org serves critical roles in G protein-coupled receptor (GPCR) and sary for central nervous system physiology and behavior. heterotrimeric G protein signal transduction. RGS proteins are Accumulating evidence has revealed key roles for specific RGS best understood as negative regulators of GPCR/G protein proteins in multiple signaling pathways at neuronal synapses, signaling. They achieve this by acting as GTPase activating regulating both pre- and postsynaptic signaling events and proteins (GAPs) for Ga subunits and accelerating the turnoff of G synaptic plasticity. Here, we review and highlight the current protein signaling. Many RGS proteins also bind additional knowledge of specific RGS proteins (RGS2, RGS4, RGS7, signaling partners that either regulate their functions or enable RGS9-2, and RGS14) that have been clearly demonstrated to them to regulate other important signaling events.