Lenzinger Berichte 94/2018
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Making! the E-Magazine for the Fibrous Forest Products Sector
PAPERmaking! The e-magazine for the Fibrous Forest Products Sector Produced by: The Paper Industry Technical Association Volume 5 / Number 1 / 2019 PAPERmaking! FROM THE PUBLISHERS OF PAPER TECHNOLOGY Volume 5, Number 1, 2019 CONTENTS: FEATURE ARTICLES: 1. Wastewater: Modelling control of an anaerobic reactor 2. Biobleaching: Enzyme bleaching of wood pulp 3. Novel Coatings: Using solutions of cellulose for coating purposes 4. Warehouse Design: Optimising design by using Augmented Reality technology 5. Analysis: Flow cytometry for analysis of polyelectrolyte complexes 6. Wood Panel: Explosion severity caused by wood dust 7. Agriwaste: Soda-AQ pulping of agriwaste in Sudan 8. New Ideas: 5 tips to help nurture new ideas 9. Driving: Driving in wet weather - problems caused by Spring showers 10. Women and Leadership: Importance of mentoring and sponsoring to leaders 11. Networking: 8 networking skills required by professionals 12. Time Management: 101 tips to boost everyday productivity 13. Report Writing: An introduction to report writing skills SUPPLIERS NEWS SECTION: Products & Services: Section 1 – PITA Corporate Members: ABB / ARCHROMA / JARSHIRE / VALMET Section 2 – Other Suppliers Materials Handling / Safety / Testing & Analysis / Miscellaneous DATA COMPILATION: Installations: Overview of equipment orders and installations since November 2018 Research Articles: Recent peer-reviewed articles from the technical paper press Technical Abstracts: Recent peer-reviewed articles from the general scientific press Events: Information on forthcoming national and international events and courses The Paper Industry Technical Association (PITA) is an independent organisation which operates for the general benefit of its members – both individual and corporate – dedicated to promoting and improving the technical and scientific knowledge of those working in the UK pulp and paper industry. -
The Effect of Organosolv Pretreatment Variables on Enzymatic Hydrolysis of Sugarcane Bagasse
Chemical Engineering Journal 168 (2011) 1157–1162 Contents lists available at ScienceDirect Chemical Engineering Journal journal homepage: www.elsevier.com/locate/cej The effect of organosolv pretreatment variables on enzymatic hydrolysis of sugarcane bagasse L. Mesa a, E. González a, C. Cara b, M. González a, E. Castro b, S.I. Mussatto c,∗ a Center of Analysis Process, Faculty of Chemistry and Pharmacy, Central University of Las Villas, Villa Clara, Cuba b Department of Chemical Environmental and Materials Engineering, Faculty of Experimental Sciences, University of Jaén, Jaén, Spain c Institute for Biotechnology and Bioengineering, Centre of Biological Engineering, University of Minho, Campus de Gualtar, 4710-057 Braga, Portugal article info abstract Article history: Sugarcane bagasse pretreated with dilute-acid was submitted to an organosolv ethanol process with Received 10 November 2010 NaOH under different operational conditions (pretreatment time, temperature, and ethanol concentra- Received in revised form 27 January 2011 tion) aiming to maximize the glucose yield in the subsequent enzymatic hydrolysis stage. The different Accepted 3 February 2011 pretreatment conditions resulted in variations in the chemical composition of the solid residue as well as in the glucose recovered by enzymatic hydrolysis. All the studied variables presented significant (p < 0.05) Keywords: influence on the process. The optimum organosolv pretreatment conditions consisted in using 30% (v/v) Sugarcane bagasse ethanol at 195 ◦C, during 60 min. Enzymatic hydrolysis of the residue then obtained produced 18.1 g/l Organosolv Ethanol glucose, correspondent to a yield of 29.1 g glucose/100 g sugarcane bagasse. The scale-up of this process, Enzymatic hydrolysis by performing the acid pretreatment in a 10-l semi-pilot reactor fed with direct steam, was success- Glucose fully performed, being obtained a glucose yield similar to that found when the acid pretreatment was performed in autoclave. -

Investigation of the Mechanical Properties of a Carbon Fibre-Reinforced Nylon Filament for 3D Printing
machines Article Investigation of the Mechanical Properties of a Carbon Fibre-Reinforced Nylon Filament for 3D Printing Flaviana Calignano 1,* , Massimo Lorusso 2 , Ignanio Roppolo 3 and Paolo Minetola 1 1 Department of Management and Production Engineering, Politecnico di Torino, Corso Duca degli Abruzzi 24, 10129 Turin, Italy; [email protected] 2 Istituto Italiano di Tecnologia, Center for Sustainable Future Technologies IIT@Polito, Corso Trento 21, 10129 Turin, Italy; [email protected] 3 Department of Applied Science and Technology, Politecnico di Torino, Corso Duca degli Abruzzi 24, 10129 Turin, Italy; [email protected] * Correspondence: fl[email protected]; Tel.: +39-011-090-7218 Received: 19 July 2020; Accepted: 2 September 2020; Published: 4 September 2020 Abstract: Additive manufacturing (i.e., 3D printing) has rapidly developed in recent years. In the recent past, many researchers have highlighted the development of in-house filaments for fused filament fabrication (FFF), which can extend the corresponding field of application. Due to the limited mechanical properties and deficient functionality of printed polymer parts, there is a need to develop printable polymer composites that exhibit high performance. This study analyses the actual mechanical characteristics of parts fabricated with a low-cost printer from a carbon fibre-reinforced nylon filament. The results show that the obtained values differ considerably from the values presented in the datasheets of various filament suppliers. Moreover, the hardness and tensile strength are influenced by the building direction, the infill percentage, and the thermal stresses, whereas the resilience is affected only by the building direction. Furthermore, the relationship between the mechanical properties and the filling factor is not linear. -

Extended Impregnation Kraft Cooking of Softwood: Effects on Reject, Yield, Pulping Uniformity, and Physical Properties
Extended Impregnation Kraft Cooking of Softwood: Effects on reject, yield, pulping uniformity, and physical properties Katarina Karlström Licentiate thesis Royal Institute of Technology (KTH) Department of Fibre and Polymer Technology Division of Wood Chemistry and Pulp Technology Stockholm 2009 TRITA-CHE-Report 2009:59 ISSN 1654-1081 ISBN 978-91-7415-496-2 Extended impregnation kraft cooking of softwood: Effects on reject, yield, pulping uniformity, and physical properties Katarina Karlström AKADEMISK AVHANDLING Som med tillstånd av Kungliga Tekniska Högskolan i Stockholm framlägges till offentlig granskning för avläggande av teknologie licentiatexamen fredagen den 18:e december 2009, kl. 10.00 i STFI-salen, Innventia AB, Drottning Kristinas väg 61, Stockholm. Avhandlingen försvaras på svenska. © Katarina Karlström Stockholm 2009 Department of Fibre and Polymer Technology Teknikringen 56-58 SE-100 44 Stockholm Sweden Abstract Converting wood into paper is a complex process involving many different stages, one of which is pulping. Pulping involves liberating the wood fibres from each other, which can be done either chemically or mechanically. This thesis focuses on the most common chemical pulping method, the kraft cooking process, and especially on a recently developed improvement of the impregnation phase, which is the first part of a kraft cook. Extended impregnation kraft cooking (EIC) technique is demonstrated to be an improvement of the kraft pulping process and provides a way to utilize softwood to a higher degree, at higher pulp yield. We demonstrate that it is possible to produce softwood ( Picea abies ) kraft pulp using a new cooking technique, resulting in a pulp that can be defibrated without inline refining at as high lignin content as 8% on wood, measured as kappa numbers above 90. -

( 12 ) United States Patent
US009902815B2 (12 ) United States Patent ( 10 ) Patent No. : US 9 ,902 , 815 B2 Tamminen et al. ( 45 ) Date of Patent: Feb . 27 , 2018 ( 54 ) FUNCTIONALIZED LIGNIN AND METHOD ( 58 ) Field of Classification Search OF PRODUCING THE SAME ??? . .. C07G 1 / 00 See application file for complete search history . @(71 ) Applicant : Teknologian tutkimuskeskus VTT , VTT (FI ) ( 56 ) References Cited U . S . PATENT DOCUMENTS @(72 ) Inventors : Tarja Tamminen , Espoo (FI ) ; Jarmo Ropponen , Espoo (FI ) ; Eva - Lena Hult , 2 ,429 , 102 A 10 / 1947 Lewis et al. Espoo ( FI) ; Kristiina Poppius- Levlin , 3 , 149 , 085 A 9 / 1964 Ball et al . Espoo (FI ) 4 ,017 ,430 A * 4 / 1977 Briggs .. .. .. 530 / 502 5 ,773 , 590 A * 6 / 1998 Hart 530 / 500 6 ,172 ,204 B1 * 1 /2001 Sarkanen et al. 530 /500 @(73 ) Assignee : Teknologian tutkimuskeskus VTT Oy, 2010 /0105798 A1 * 4 /2010 Hasegawa . .. .. 522 / 99 Espoo (FI ) 2010 /0152428 A1* 6 / 2010 Gifford et al . .. .. .. 530 / 504 2010 / 0204368 A1 * 8 / 2010 Benko et al. .. .. .. .. .. .. .. 524 / 73 ( * ) Notice : Subject to any disclaimer, the term of this 2010 / 0331531 A1 * 12 / 2010 Mykytka .. .. 530 / 501 patent is extended or adjusted under 35 2011/ 0263836 A1 * 10 / 2011 Vuorenpalo et al. .. .. 530 / 500 U . S . C . 154 ( b ) by 33 days. 2012 /0012035 A1 * 1 / 2012 Blank et al. .. .. 106 / 802 ( 21) Appl. No. : 14 /348 , 904 OTHER PUBLICATIONS (22 ) PCT Filed : Oct. 8 , 2012 Holmbom ( Journal of the American Oil Chemists Society , 1977 , p . 289 - 293 ) . * Stenberg et al . (Surface Coatings International Part B : Coatings ( 86 ) PCT No. : PCT/ FI2012 /050965 Transactions , vol. 88 , B2 , 83 - 156 , 2005 ) . -

What Happens to Cellulosic Fibers During Papermaking and Recycling? a Review
PEER-REVIEWED REVIEW ARTICLE ncsu.edu/bioresources WHAT HAPPENS TO CELLULOSIC FIBERS DURING PAPERMAKING AND RECYCLING? A REVIEW Martin A. Hubbe,* Richard A. Venditti, and Orlando J. Rojas Both reversible and irreversible changes take place as cellulosic fibers are manufactured into paper products one or more times. This review considers both physical and chemical changes. It is proposed that by understanding these changes one can make better use of cellulosic fibers at various stages of their life cycles, achieving a broad range of paper performance characteristics. Some of the changes that occur as a result of recycling are inherent to the fibers themselves. Other changes may result from the presence of various contaminants associated with the fibers as a result of manufacturing processes and uses. The former category includes an expected loss of swelling ability and decreased wet-flexibility, especially after kraft fibers are dried. The latter category includes effects of inks, de-inking agents, stickies, and additives used during previous cycles of papermaking. Keywords: Paper recycling, Drying, Deinking, Hornification, Inter-fiber bonding, Refining, Fines, Fiber length, Conformability Contact information: Department of Forest Biomaterials Science and Engineering, Box 8005, North Carolina State University, Raleigh, NC 27695-8005, USA; *Corresponding author: [email protected] INTRODUCTION Cellulosic fibers can change significantly when formed into a wet web of paper and subsequently subjected to such processes as pressing, drying, printing, storage, repulping, and deinking. Some of the changes can be subtle. Often it is possible to substitute recovered fibers in place of virgin fibers used for the production of paper or paperboard. On the other hand, characteristic differences between recycled fibers and virgin fibers (fresh from pulping wood, not recycled) can be expected; many of these can be attributed to drying. -

Deinking of Screen-Printed Electrodes Printed on Invasive Plant-Based Paper
sustainability Article Article DeinkingDeinking of of Screen-Printed Screen-Printed Electrodes Printed on InvasiveInvasive Plant-Based Plant-Based Paper UrškaUrška Kav Kavˇciˇc*čič *, Igor, Igor Karlovits Karlovits and and Janja Janja Zule Zule PulpPulp and and Paper Paper Institute, Institute, Bogiši Bogiši´ceva8,ćeva 8, 1000 Ljubljana, Slov Slovenia;enia; igor.karlovits@icp-lj. [email protected] (I.K.); janja.zule@icp- [email protected] (J.Z.) (J.Z.) * Correspondence: [email protected] Received: 21 January 2020; Accepted: 6 February 2020; Published: date Received: 21 January 2020; Accepted: 9 February 2020; Published: 12 February 2020 Abstract: The deinking of paper-based printed electronics is a growing concern regarding the Abstract: The deinking of paper-based printed electronics is a growing concern regarding the increase increase of printed electronics products. The benefits of using paper-based substrates instead of of printed electronics products. The benefits of using paper-based substrates instead of polymer polymer or ceramic for the single-use printed electrodes can contribute to sustainability goals. The or ceramic for the single-use printed electrodes can contribute to sustainability goals. The use of use of invasive plant species for making paper substrates for printed electronics is a unique invasive plant species for making paper substrates for printed electronics is a unique opportunity opportunity to have several environmental benefits. In this study, the recycling issue of these to have several environmental benefits. In this study, the recycling issue of these products through products through the use of the deinking technique was evaluated. Screen-printed electrodes the use of the deinking technique was evaluated. -

Fungal Pretreatment for Organosolv Pulping and Dissolving Pulp Production
13 Fungal Pretreatment for Organosolv Pulping and Dissolving Pulp Production ANDRÉ FERRAZ, LYUDMIL CHRISTOV, AND MASOOD AKHTAR INTRODUCTION Biopulping is the fungal pretreatment of wood chips, designed as a solid-state fer- mentation process, for production of mechanical or chemical pulps (cf. Chapters 10 and 12). The concept of biopulping is based on the ability of some white-rot fungi to colonize and degrade selectively the lignin in wood, thereby leaving the cellulose relatively intact. This chapter presents an evaluation of biopulping and biobleaching with white-rot fungi as pretreatment steps in organosolv pulping and in dissolving pulp production. Fungal-organosolv pulping is focused mainly on chemical and topo- chemical bases for the combination of biopulping and organosolv processes. Experi- mental data are presented to illustrate the potential of this fungal-chemical method to produce wood pulps. Biopulping and biobleaching with selected white-rot fungi are also evaluated as potential methods for production of dissolving pulp. A report in part has been submitted for publication in Enzyme and Microbial Technology. This chapter is based mainly on the work from a collaborative project between the Department of Microbiology and Biochemistry, The University of Orange Free State, of Bloemfontein South Africa and the USDA Forest Products Laboratory in Madison, Wisconsin. Fungal-organosolv pulping methods are based mainly on the work developed at the Department of Biotechnology in Lorena, Brazil, and the Renewable Resource Laboratory in Concepcion, Chile. ORGANOSOLV PULPING Background The need to find new technologies for pulp and paper production that cause less environmental impact has contributed to an increase in the number of studies on Environmentally Friendly Technologies for the Pulp and Paper Industry, edited by Raymond A. -

Value Chains for Production of Renewable Transportation Fuels Using Intermediates - Case Studies for Bio-Oils in Refinery Applications and Bio-Sng
REPORT f3 2016:05 VALUE CHAINS FOR PRODUCTION OF RENEWABLE TRANSPORTATION FUELS USING INTERMEDIATES - CASE STUDIES FOR BIO-OILS IN REFINERY APPLICATIONS AND BIO-SNG Report from an f3 project June 2016 Authors: Marie Anheden, Christian Ehn & Valeria Lundberg, INNVENTIA AB Karin Pettersson, Chalmers University of Technology Malin Fugelsang & Carl Johan Hjerpe, ÅF Industri AB Åsa Håkansson, Preem AB Ingemar Gunnarsson, Göteborg Energi AB VALUE CHAINS FOR PRODUCTION OF RENEWABLE TRANSPORTATION FUELS USING INTERMEDIATES PREFACE This report is the result of a collaborative project within the Swedish Knowledge Centre for Renewa- ble Transportation Fuels (f3). f3 is a networking organization, which focuses on development of envi- ronmentally, economically and socially sustainable renewable fuels, and Provides a broad, scientifically based and trustworthy source of knowledge for industry, gov- ernments and public authorities, Carries through system oriented research related to the entire renewable fuels value chain, Acts as national platform stimulating interaction nationally and internationally. f3 partners include Sweden’s most active universities and research institutes within the field, as well as a broad range of industry companies with high relevance. f3 has no political agenda and does not con- duct lobbying activities for specific fuels or systems, nor for the f3 partners’ respective areas of inter- est. The f3 centre is financed jointly by the centre partners, the Swedish Energy Agency and the region of Västra Götaland. f3 also receives funding from Vinnova (Sweden’s innovation agency) as a Swedish advocacy platform towards Horizon 2020. Chalmers Industriteknik (CIT) functions as the host of the f3 organization (see www.f3centre.se). The project is financed and carried out within the f3 - Energimyndigheten (Swedish Energy Agency) collaborative research program “Förnybara drivmedel och system” (Renewable transportation fuels and systems). -

Natural Fibers and Fiber-Based Materials in Biorefineries
Natural Fibers and Fiber-based Materials in Biorefineries Status Report 2018 This report was issued on behalf of IEA Bioenergy Task 42. It provides an overview of various fiber sources, their properties and their relevance in biorefineries. Their status in the scientific literature and market aspects are discussed. The report provides information for a broader audience about opportunities to sustainably add value to biorefineries by considerin g fiber applications as possible alternatives to other usage paths. IEA Bioenergy Task 42: December 2018 Natural Fibers and Fiber-based Materials in Biorefineries Status Report 2018 Report prepared by Julia Wenger, Tobias Stern, Josef-Peter Schöggl (University of Graz), René van Ree (Wageningen Food and Bio-based Research), Ugo De Corato, Isabella De Bari (ENEA), Geoff Bell (Microbiogen Australia Pty Ltd.), Heinz Stichnothe (Thünen Institute) With input from Jan van Dam, Martien van den Oever (Wageningen Food and Bio-based Research), Julia Graf (University of Graz), Henning Jørgensen (University of Copenhagen), Karin Fackler (Lenzing AG), Nicoletta Ravasio (CNR-ISTM), Michael Mandl (tbw research GesmbH), Borislava Kostova (formerly: U.S. Department of Energy) and many NTLs of IEA Bioenergy Task 42 in various discussions Disclaimer Whilst the information in this publication is derived from reliable sources, and reasonable care has been taken in its compilation, IEA Bioenergy, its Task42 Biorefinery and the authors of the publication cannot make any representation of warranty, expressed or implied, regarding the verity, accuracy, adequacy, or completeness of the information contained herein. IEA Bioenergy, its Task42 Biorefinery and the authors do not accept any liability towards the readers and users of the publication for any inaccuracy, error, or omission, regardless of the cause, or any damages resulting therefrom. -
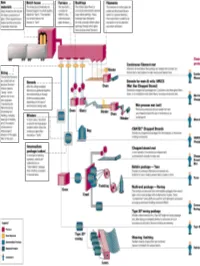
All About Fibers
RawRaw MaterialsMaterials ¾ More than half the mix is silica sand, the basic building block of any glass. ¾ Other ingredients are borates and trace amounts of specialty chemicals. Return © 2003, P. Joyce BatchBatch HouseHouse && FurnaceFurnace ¾ The materials are blended together in a bulk quantity, called the "batch." ¾ The blended mix is then fed into the furnace or "tank." ¾ The temperature is so high that the sand and other ingredients dissolve into molten glass. Return © 2003, P. Joyce BushingsBushings ¾The molten glass flows to numerous high heat-resistant platinum trays which have thousands of small, precisely drilled tubular openings, called "bushings." Return © 2003, P. Joyce FilamentsFilaments ¾This thin stream of molten glass is pulled and attenuated (drawn down) to a precise diameter, then quenched or cooled by air and water to fix this diameter and create a filament. Return © 2003, P. Joyce SizingSizing ¾The hair-like filaments are coated with an aqueous chemical mixture called a "sizing," which serves two main purposes: 1) protecting the filaments from each other during processing and handling, and 2) ensuring good adhesion of the glass fiber to the resin. Return © 2003, P. Joyce WindersWinders ¾ In most cases, the strand is wound onto high-speed winders which collect the continuous fiber glass into balls or "doffs.“ Single end roving ¾ Most of these packages are shipped directly to customers for such processes as pultrusion and filament winding. ¾ Doffs are heated in an oven to dry the chemical sizing. Return © 2003, P. Joyce IntermediateIntermediate PackagePackage ¾ In one type of winding operation, strands are collected into an "intermediate" package that is further processed in one of several ways. -

Carbon Fiber Supply and Demand
Carbon Fiber Supply and Demand Supply of PAN based carbon fiber New players could make all the difference San Diego, California, USA October 23 2007 The ongoing shortage of PAN based carbon fibers has seen the planning, development and realization of new carbon fiber manufacturing capacity in new countries. The start up of new plant faces many hurdles, especially in the face of competition that has been in the business for many decades. However these hurdles have been overcome and this process will enable carbon fiber to become truly a commodity product, available to the vast range of current and new composite applications. Latest News (Press releases in the last 4 weeks) Supply October 11, 2007 Capacity Expansion of Carbon Fiber “TENAX®” Toho Tenax Co., Ltd. (Head Office: Bunkyo-ku, Tokyo, President: Yoshikuni Utsunomiya), which is engaged in the business of carbon fibers within the Teijin Group, has decided to add a new carbon fiber production line at Toho Tenax Europe GmbH (located at Wuppertal, Germany. October 05, 2007 SGL Group plans to triple carbon fiber capacities SGL Group plans to triple carbon fiber capacities Capacity increase up to 12,000 metric tons by 2012 Demand September 27, 2007 British Airways Green light for aircraft orders British Airways has today placed an order for 12 Airbus A380 and 24 Boeing 787 aircraft with options for a further seven Airbus A380s and18 Boeing 787s. The British Airways Order The new aircraft will replace 34 of the airline's long haul fleet and will be delivered between 2010 and 2014. The order, including options, will give the airline the ability to grow its capacity by up to four per cent per year and the flexibility to tailor its future capacity growth in line with market conditions.