Urey (1947): Thermodynamic Properties of Isotope Substances
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

The Dole Effect and Its Variations During the Last 130,000 Years As Measured in the Vostok Ice Core
GLOBAL BIOGEOCHEMICAL CYCLES, VOL. 8, NO. 3, PAGES 363-376, SEPTEMBER 1994 The Dole effect and its variations during the last 130,000 years as measured in the Vostok ice core Michael Bender • and Todd Sowers 2 GraduateSchool of Oceanography,University of RhodeIsland, Kingston Laurent Labeyrie Centredes Faibles Radioactivites, Centre National de la RechercheScientifique, Commissariat a L'EnergieAtomique, Gif-sur- Yvette, France Abstract. We review the currentunderstanding of the Dole effect (the observeddifference between the •5180of atmospheric0 2 andthat of seawater)and its causes, extend the record of variationsin theDole effectback to 130kyr before present using data on the •5180 of 0 2obtained from studying the Vostok ice core(Sowers et al., 1993), anddiscuss the significanceof temporalvariations. The Dole effectreflects oxygenisotope fractionation during photosynthesis, respiration, and hydrologic processes (evaporation, precipitation,and evapotranspiration). Our bestprediction of the present-dayDole effect,+20.8 %0,is considerablylower thanthe observedvalue, + 23.5 %0,and we discusspossible causes of this discrepancy. During the past 130 kyr, the Dole effecthas been 0.05 %0lower thanthe present value, on average.The standarddeviation of the Dole effect from the meanhas been only _+0.2 %0,and the Dole effect is nearly unchangedbetween glacial maxima and interglacial periods. The smallvariability in theDole effect suggeststhat relative rates of primaryproduction in theland and marine realms have been relatively constant.Most periodicvariability in the Dole effectis in the precessionband, suggesting that changes in thisglobal biogeochemical term reflects variations in low-latitudeland hydrology and productivity or possiblyvariability in low-latitudeoceanic productivity. Introduction they can potentially be made to do so. Ice core reconstructionsof the concentrationsof bioactivegases in air Our understanding of the response of the biosphere to provide an integratedglobal signal which complementsthe Pleistoceneclimate change is actually quite limited. -

Compilation of Minimum and Maximum Isotope Ratios of Selected Elements in Naturally Occurring Terrestrial Materials and Reagents by T
U.S. Department of the Interior U.S. Geological Survey Compilation of Minimum and Maximum Isotope Ratios of Selected Elements in Naturally Occurring Terrestrial Materials and Reagents by T. B. Coplen', J. A. Hopple', J. K. Bohlke', H. S. Reiser', S. E. Rieder', H. R. o *< A R 1 fi Krouse , K. J. R. Rosman , T. Ding , R. D. Vocke, Jr., K. M. Revesz, A. Lamberty, P. Taylor, and P. De Bievre6 'U.S. Geological Survey, 431 National Center, Reston, Virginia 20192, USA 2The University of Calgary, Calgary, Alberta T2N 1N4, Canada 3Curtin University of Technology, Perth, Western Australia, 6001, Australia Institute of Mineral Deposits, Chinese Academy of Geological Sciences, Beijing, 100037, China National Institute of Standards and Technology, 100 Bureau Drive, Stop 8391, Gaithersburg, Maryland 20899 "institute for Reference Materials and Measurements, Commission of the European Communities Joint Research Centre, B-2440 Geel, Belgium Water-Resources Investigations Report 01-4222 Reston, Virginia 2002 U.S. Department of the Interior GALE A. NORTON, Secretary U.S. Geological Survey Charles G. Groat, Director The use of trade, brand, or product names in this report is for identification purposes only and does not imply endorsement by the U.S. Government. For additional information contact: Chief, Isotope Fractionation Project U.S. Geological Survey Mail Stop 431 - National Center Reston, Virginia 20192 Copies of this report can be purchased from: U.S. Geological Survey Branch of Information Services Box 25286 Denver, CO 80225-0286 CONTENTS Abstract...................................................... -
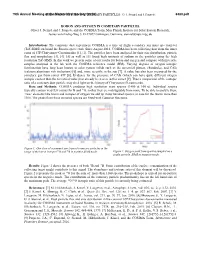
Boron and Oxygen in Cometary Particles: O. J
79th Annual Meeting ofBORON the Meteoritical AND OXYGEN Society IN COMETARY (2016) PARTICLES: O. J. Stenzel and J. Paquette 6380.pdf BORON AND OXYGEN IN COMETARY PARTICLES. Oliver J. Stenzel and J. Paquette and the COSIMA Team, Max Planck Institute for Solar System Research, Justus-von-Liebig-Weg 3, D-37077 Göttingen, Germany, [email protected]. Introduction: The cometary dust experiment COSIMA is a time of flight secondary ion mass spectrometer (ToF-SIMS) on board the Rosetta space craft. Since August 2014, COSIMA has been collecting dust from the inner coma of 67P/Churyumov-Gerasimenko [1], [2]. The particles have been analyzed for their size distribution, particle flux and morphology [3], [4]. [5] as well as [1] found high amounts of sodium in the particles using the high resolution ToF-SIMS. In this work we present some of our results for boron and oxygen and compare with meteorite samples analyzed in the lab with the COSIMA reference model (RM). Varying degrees of oxygen isotopic fractionation have long been known in solar system solids such as the terrestrial planets, chondrules, and CAIs (calcium-aluminum-rich inclusions) [6] and, more recently, in the sun [7]. A value has also been measured for the cometary gas from comet 67P [8]. Evidence for the presence of CAIs (which can have quite different oxygen isotopic content than the terrestrial value) has already been seen in this comet [9]. Thus a comparison of the isotopic ratio of a cometary dust particle may shed light on the history of Churyumov-Gerasimenko. Data and Methods: COSIMA produces high resolution mass spectra (1400 at 100 u). -

Investigation of Nitrogen Cycling Using Stable Nitrogen and Oxygen Isotopes in Narragansett Bay, Ri
University of Rhode Island DigitalCommons@URI Open Access Dissertations 2014 INVESTIGATION OF NITROGEN CYCLING USING STABLE NITROGEN AND OXYGEN ISOTOPES IN NARRAGANSETT BAY, RI Courtney Elizabeth Schmidt University of Rhode Island, [email protected] Follow this and additional works at: https://digitalcommons.uri.edu/oa_diss Recommended Citation Schmidt, Courtney Elizabeth, "INVESTIGATION OF NITROGEN CYCLING USING STABLE NITROGEN AND OXYGEN ISOTOPES IN NARRAGANSETT BAY, RI" (2014). Open Access Dissertations. Paper 209. https://digitalcommons.uri.edu/oa_diss/209 This Dissertation is brought to you for free and open access by DigitalCommons@URI. It has been accepted for inclusion in Open Access Dissertations by an authorized administrator of DigitalCommons@URI. For more information, please contact [email protected]. INVESTIGATION OF NITROGEN CYCLING USING STABLE NITROGEN AND OXYGEN ISOTOPES IN NARRAGANSETT BAY, RI BY COURTNEY ELIZABETH SCHMIDT A DISSERTATION SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY IN OCEANOGRAPHY UNIVERSITY OF RHODE ISLAND 2014 DOCTOR OF PHILOSOPHY DISSERTATION OF COURTNEY ELIZABETH SCHMIDT APPROVED: Thesis Committee: Major Professor Rebecca Robinson Candace Oviatt Arthur Gold Nassar H. Zawia DEAN OF THE GRADUATE SCHOOL UNIVERSITY OF RHODE ISLAND 2014 ABSTRACT Estuaries regulate nitrogen (N) fluxes transported from land to the open ocean through uptake and denitrification. In Narragansett Bay, anthropogenic N loading has increased over the last century with evidence for eutrophication in some regions of Narragansett Bay. Increased concerns over eutrophication prompted upgrades at wastewater treatment facilities (WWTFs) to decrease the amount of nitrogen discharged. The upgrade to tertiary treatment – where bioavailable nitrogen is reduced and removed through denitrification – has occurred at multiple facilities throughout Narragansett Bay’s watershed. -

Analytical Developments in the Measurements of Boron, Nitrate
XA9848324 ANALYTICAL DEVELOPMENTS IN THE MEASUREMENTS OF BORON, NITRATE, PHOSPHATE AND SULPHATE ISOTOPES AND CASE EXAMPLES OF DISCRIMINATION OF NITROGEN AND SULPHUR SOURCES IN POLLUTION STUDIES J. AGGARWAL, D.S. SHEPPARD Institute of Geological & Nuclear Sciences, Lower Hutt B.W. ROBINSON Wairakei Research Centre, Institute of Geological & Nuclear Sciences, Taupo New Zealand Abstract - Methods are documented for the analysis of B isotopes, O and N isotopes in nitrates. B isotopes can be measured by negative ion thermal ionisation mass spectrometry. Nitrate is recovered from groundwaters by ion exchange and the resulting silver nitrate combusted for stable isotope gas analysis. Oxygen isotope analysis of phosphates can be determined by generating and analysing CO2 gas from the combustion of silver phosphate produced from aqueous samples. Sulphate in ground and surface waters can be separated and concentrated by ion exchange and precipitated as barium sulphate. This is reacted with graphite to yield CO2 and CO, the latter being spark discharged to CO2 and the total CO2 measured for oxygen isotope analysis. Barium sulphide from this reaction is converted to silver sulphide which is reacted with cuprous oxide to give SO2 gas for sulphur isotope measurements. A case study of the semi-rural Manakau area in New Zealand was conducted to see if nitrate isotopes could be used to detect the source of nitrate contamination (groundwater nitrate <0.5 to 11.3 mg/L NO'3-N). Nitrogen isotope (+4 to +12%o) coupled with oxygen isotope measurements (+5 to +9%o) demonstrated that the nitrogen is not sourced from fertilisers but from some combination of septic tank and animal waste. -

Isotopes Are Atoms with the Same # of Protons (Making Them the SAME ELEMENT), but Have a Different # of Neutrons
Each element has a unique # of protons. So…EVERY SINGLE ATOM of that element MUST HAVE the SAME number of protons, ALWAYS! This is the reason why each element has a unique Atomic Number that never changes! Isotopes are atoms with the same # of protons (making them the SAME ELEMENT), but have a different # of neutrons. Having different # of neutrons for the same element means that the isotopes will have different mass numbers, too! There are 3 isotopes of oxygen: 8 8 8 O8 O9 O10 neutronsOxygen neutronsOxygen neutronsOxygen ALL oxygen16 isotopes 17have an atomic 18# of 8, so they all have 8 protons! How many protons does each isotope have? How many neutrons does each isotope have? There are 3 isotopes of carbon: 6 6 6 C6 C7 C8 neutronsCarbon neutronsCarbon neutronsCarbon ALL carbon12 isotopes 13have an atomic #14 of 6, so they all have 6 protons! How many protons does each isotope have? How many neutrons does each isotope have? Atomic Masses on the periodic table are normally decimals. WHY?? Atomic Masses are decimals because it is an AVERAGE mass of all of the element’s isotopes. Some isotopes are more common than others, so this is a weighted average. 5 So the Mass # from the periodic table is B always closest to the most common Boron isotope’s mass. 10.81 In the universe, there are MUCH more carbon-12 atoms that exist than carbon-13 and carbon-14 atoms. 6 C Carbon- 12 Carbon 12.01 When you find the weighted average of the masses of the carbon isotopes, the average will be closest to 12! The Atomic Mass of calcium is 40.08. -

Oxygen Isotopes in Nature and in the Laboratory Author(S): Harold C
Oxygen Isotopes in Nature and in the Laboratory Author(s): Harold C. Urey Source: Science, New Series, Vol. 108, No. 2810 (Nov. 5, 1948), pp. 489-496 Published by: American Association for the Advancement of Science Stable URL: http://www.jstor.org/stable/1677444 Accessed: 22/10/2008 09:24 Your use of the JSTOR archive indicates your acceptance of JSTOR's Terms and Conditions of Use, available at http://www.jstor.org/page/info/about/policies/terms.jsp. JSTOR's Terms and Conditions of Use provides, in part, that unless you have obtained prior permission, you may not download an entire issue of a journal or multiple copies of articles, and you may use content in the JSTOR archive only for your personal, non-commercial use. Please contact the publisher regarding any further use of this work. Publisher contact information may be obtained at http://www.jstor.org/action/showPublisher?publisherCode=aaas. Each copy of any part of a JSTOR transmission must contain the same copyright notice that appears on the screen or printed page of such transmission. JSTOR is a not-for-profit organization founded in 1995 to build trusted digital archives for scholarship. We work with the scholarly community to preserve their work and the materials they rely upon, and to build a common research platform that promotes the discovery and use of these resources. For more information about JSTOR, please contact [email protected]. American Association for the Advancement of Science is collaborating with JSTOR to digitize, preserve and extend access to Science. http://www.jstor.org Oxygen Isotopes in Nature and in the Laboratory Harold C. -

The Atmospheric Oxygen Cycle: the Oxygen Isotopes of Atmospheric C02 and 02 and the 02/N2 Ratio
REVIEWS OF GEOPHYSICS, SUPPLEMENT, PAGES 1253-1262, JULY 1995 U.S. NATIONAL REPORT TO INTERNATIONAL UNION OF GEODESY AND GEOPHYSICS 1991-1994 The atmospheric oxygen cycle: The oxygen isotopes of atmospheric C02 and 02 and the 02/N2 ratio Ralph F. Keeling Scripps Institution of Oceanography, University of California, San Diego, La Jolla Introduction to come to isotopic equilibrium with liquid water is the same as the time scale for hydration, i.e., around 30 sec 18 16 Oxygen is the most abundant element in the earth's onds [Mills and Urey, 1940]. The 0/ 0 ratio of C02 crust, it accounts for 89% of the mass of the ocean, and in equilibrium with water at 25° C is 1.041 times higher 18 16 it is the second most abundant element in the earth's than the 0/ 0 ratio of the water. This equilibrium atmosphere. Much work on the oxygen cycle has fo fractionation factor varies slightly with temperature. Isotopic ratios are generally reported according to cused on the question of the origin of atmospheric 02 and its variations over geologic time [see Kump et al, 18 16 18 16 1991, and references therein). This review focuses on S = ( 0/ 0)sampie/( 0/ 0),Wflrd " 1 (1) several other aspects of the oxygen cycle including the short-term controls on the oxygen isotopic abundance where the S value is customarily multiplied by 1000 and expressed in per mille (%<>)• Bottinga and Craig [1969] of atmospheric C02 and 02, and the short-term vari- abilitiy in the 0 /N ratio. -

Exchange of Oxygen and Hydrogen Isotopes Between Water and Water Vapor: Experimental Results
UNLV Retrospective Theses & Dissertations 1-1-1993 Exchange of oxygen and hydrogen isotopes between water and water vapor: Experimental results Michael Willem Slattery University of Nevada, Las Vegas Follow this and additional works at: https://digitalscholarship.unlv.edu/rtds Repository Citation Slattery, Michael Willem, "Exchange of oxygen and hydrogen isotopes between water and water vapor: Experimental results" (1993). UNLV Retrospective Theses & Dissertations. 295. http://dx.doi.org/10.25669/z6nr-dvzi This Thesis is protected by copyright and/or related rights. It has been brought to you by Digital Scholarship@UNLV with permission from the rights-holder(s). You are free to use this Thesis in any way that is permitted by the copyright and related rights legislation that applies to your use. For other uses you need to obtain permission from the rights-holder(s) directly, unless additional rights are indicated by a Creative Commons license in the record and/ or on the work itself. This Thesis has been accepted for inclusion in UNLV Retrospective Theses & Dissertations by an authorized administrator of Digital Scholarship@UNLV. For more information, please contact [email protected]. INFORMATION TO USERS This manuscript has been reproduced from the microfilm master. UMI films the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter face, while others may be from any type of computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely affect reproduction. -

Isotope Geology Without the Invention of the Mass Spectrometer.This Was the Major Contribution of Thomson (1914)Andaston (1918)
CHAPTER ONE IsotopesIsotopes and radioactivityradioactivity 1.1 Reminders about the atomic nucleus Inthe model ¢rstdeveloped byNiels Bohrand Ernest Rutherford and extended byArnold Sommerfeld, the atom is composed of two entities: a central nucleus, containing most of the mass, and an array oforbiting electrons.1 The nucleus carries a positive charge of þZe, which is balanced by the electron cloud’s negative charge of ÀZe.The number of protons, Z, is matchedin an electrically neutral atomby thenumberofelectrons. Eachoftheseparti- cles carries anegative electric chargee. As a rough description, the nucleus of any element is made up of two types of particle, neutrons and protons. A neutron is slightly heavier than a proton with a mass of À27 À27 mn ¼ 1.674 95 Á 10 kg compared with mp ¼ 1.672 65 Á 10 kg for the proton.While of similar masses, then, the two particles di¡er above all in their charges. The proton has a positive charge (þ e) while the neutron is electrically neutral.The number of protons (Z)is the atomic number.ThesumA ¼ N þ Z of the number of neutrons (N) plus the number of protons (Z) gives the mass number. This provides a measure of the mass of the nuclide in question if we take as our unit the approximate mass of the neutron or proton. Thomson (1914)andAston(1919)showedthat,foragivenatomicnumberZ,thatis,foragivenposition in Mendeleyev’s periodic table, there are atoms with di¡erent mass numbers A,andthere- fore nuclei which di¡er in the number of neutrons they contain (see Plate1). Such nuclides areknown as the isotopesofan element. -

Biogenic Carbonates – Oxygen
Chapter 6 BIOGENIC CARBONATES – OXYGEN Contents 6.1 Introduction ................................................................................................................... 1 6.2 The Phosphoric Acid Method ....................................................................................... 2 6.2.1 A major breakthrough ............................................................................................ 2 6.2.2 Acid Fractionation Factors ..................................................................................... 3 6.2.3. Applicability ......................................................................................................... 5 6.3 The Oxygen Isotope Paleotemperature Scale ............................................................... 6 6.4 Factors Affecting Oxygen Isotope Paleotemperatures ............................................... 11 6.4.1 Variations in 18O of ocean water in space and time .......................................... 11 6.4.2 Vital effects .......................................................................................................... 13 6.4.3. Diagenesis ........................................................................................................... 14 6.4.4 Ecology of the organism ...................................................................................... 19 6.5 Applications of Oxygen Isotope Paleothermometry ................................................... 19 6.5.1 The Quaternary ................................................................................................... -

Use of Stable Isotopes of Carbon, Nitrogen, and Sulfur to Identify Sources of Nitrogen in Surface Waters in the Lower Susquehanna River Basin, Pennsylvania
Use of Stable Isotopes of Carbon, Nitrogen, and Sulfur to Identify Sources of Nitrogen in Surface Waters in the Lower Susquehanna River Basin, Pennsylvania By CHARLES A. CRAVOTTA III Prepared in cooperation with the PENNSYLVANIA DEPARTMENT OF ENVIRONMENTAL PROTECTION U.S. GEOLOGICAL SURVEY WATER-SUPPLY PAPER 2497 U.S. DEPARTMENT OF THE INTERIOR GALE A. NORTON, Secretary U.S. GEOLOGICAL SURVEY Charles G. Groat, Director Any use of trade, product, or firm names in this publication is for descriptive purposes only and does not imply endorsement by the U.S. Government. For sale by the U.S. Geological Survey, Branch of Information Services Box 25286 Denver, CO 80225-0286 Library of Congress Cataloging-in-Publication Data Cravotta, Charles A. Use of stable isotopes of carbon, nitrogen, and sulfur to identify sources of nitrogen in surface waters in the lower Susquehanna River Basin, Pennsylvania / by Charles A. Cravotta III. p. cm. -- (U.S. Geological Survey water-supply paper : 2497) “Prepared in cooperation with the Pennsylvania Department of Environmental Protection, Bureau of Soil and Water Conservation.” Includes bibliographic references (p. - ). Supt. of Docs. no.: I 19.13:W2497 1. Nitrogen--Environmental aspects--Susquehanna River Watershed. 2. Water--Pollution--Susquehanna River Watershed--Point source identification. 3. Radioactive tracers in water pollution research-Susquehanna River Water- shed. I. Pennsylvania. Bureau of Soil and Water Conservation. II. Title. III. Series. TD427.N5C73 1997 97-10381 628.1'61--dc21 CIP ISBN 0-607-87207-1