Analytical Developments in the Measurements of Boron, Nitrate
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Consumer Superbrands 2019 Top 10 Consumer Superbrands Relevancy
Consumer Superbrands 2019 Top 10 Consumer Superbrands BRAND CATEGORY LEGO 1 Child Products - Toys and Education Apple 2 Technology - General Gillette 3 Toiletries - Men's Grooming Rolex 4 Watches British Airways 5 Travel - Airlines Coca-Cola 6 Drinks - Non-Alcoholic - Carbonated Soft Drinks Andrex 7 Household - Kitchen Rolls, Toilet Roll and Tissues Mastercard 8 Financial - General Visa 9 Financial - General Dyson 10 Household & Personal Care Appliances Relevancy Index Top 20 BRAND CATEGORY Amazon 1 Retail - Entertainment & Gifts Aldi 2 Retail - Food & Drink Macmillan Cancer Support 3 Charities Netflix 4 Media - TV Google 5 Social, Search & Comparison Sites Lidl 6 Retail - Food & Drink PayPal 7 Financial - General LEGO 8 Child Products - Toys and Education Samsung 9 Technology - General YouTube 10 Social, Search & Comparison Sites Visa 11 Financial - General Heathrow 12 Travel - Airports Purplebricks 13 Real Estate Cancer Research UK 14 Charities Oral-B 15 Toiletries - Oral Care Apple 16 Technology - General Dyson 17 Household & Personal Care Appliances TripAdvisor 18 Travel - Agents & Tour Operators Nike 19 Sportswear & Equipment Disney 20 Child Products - Toys and Education continues... Consumer Superbrands 2019 Category Winners CATEGORY BRAND Automotive - Products Michelin Automotive - Services AA Automotive - Vehicle Manufacturer Mercedes-Benz Charities Cancer Research UK Child Products - Buggies, Seats and Cots Mamas & Papas Child Products - General JOHNSON'S Child Products - Toys and Education LEGO Drinks - Alcoholic - Beer, Ale -

Product & Service Guide
Product & Service Guide Your complete guide to JohnsonDiversey cleaning and hygiene solutions www.johnsondiversey.co.uk 2 Your complete guide to 1 JohnsonDiversey cleaning and hygiene solutions Whether you are a new or existing customer we want you to get the most from this guide. Contents Contents Features of this guide: There is a product index at the back Sustainability 2 72 Product Index A A Cif Wood Floor Cleaner 65 Enhance Foam Shampoo 27 Supply Chain 4 Agressor 31 CLAX 100 OB 48 Enhance Spot & Stain 27 Aquamat 10 56 CLAX 100 S 48 Ensign 360/460 55 Aquamat 20 56 CLAX 500 49 Ensign SM1/2 55 Aquamat 30 56 CLAX Bright 47 Ensign Stealth 1/2 55 Aquamat 45 56 CLAX Build 48 Ergodisc 1200 57 Customer Service Commitment 5 CLAX Diamond 47 Ergodisc 165 56 CLAX Elegant 3CL2 47 Ergodisc 200 56 B B CLAX Hypo 48 Ergodisc 238 56 Bactosol Beerline Cleaner 11 CLAX Kombi Citric 48 Ergodisc 400 56 Product Index Bactosol Cabinet Detergent 11 CLAX Mild 3RL1 47 Ergodisc 438 57 Bactosol Cabinet Glasswash Rinse Aid 11 CLAX Novix 49 Ergodisc Accessories 60 Kitchen Hygiene 6 Bactosol Glass Renovator 11 CLAX Oxy 4EP1 49 Ergodisc duo 56 Bactosol Hand Glasswashing Liquid 11 CLAX Perfect 48 Ergodisc Foam Generator 56 Balimat 45 58 CLAX Profi 47 Ergodisc Mini 56 Bourne Aqua Seal 27 CLAX Revita 49 Ergodisc omni 57 Bourne Seal 27 Clax Revoflow 45 Exact System 40 Bar & Cellar Cleaning 11 Bourne Traffic Liquid Wax 27 CLAX Saturn 49 Brillo Catering Scourers No.96 69 CLAX Sigma 48 Brillo Cleaner & Degreaser 68 CLAX Silver 48 F B Florzip Sweeping System 54 Brillo Concentrated -

The Dole Effect and Its Variations During the Last 130,000 Years As Measured in the Vostok Ice Core
GLOBAL BIOGEOCHEMICAL CYCLES, VOL. 8, NO. 3, PAGES 363-376, SEPTEMBER 1994 The Dole effect and its variations during the last 130,000 years as measured in the Vostok ice core Michael Bender • and Todd Sowers 2 GraduateSchool of Oceanography,University of RhodeIsland, Kingston Laurent Labeyrie Centredes Faibles Radioactivites, Centre National de la RechercheScientifique, Commissariat a L'EnergieAtomique, Gif-sur- Yvette, France Abstract. We review the currentunderstanding of the Dole effect (the observeddifference between the •5180of atmospheric0 2 andthat of seawater)and its causes, extend the record of variationsin theDole effectback to 130kyr before present using data on the •5180 of 0 2obtained from studying the Vostok ice core(Sowers et al., 1993), anddiscuss the significanceof temporalvariations. The Dole effectreflects oxygenisotope fractionation during photosynthesis, respiration, and hydrologic processes (evaporation, precipitation,and evapotranspiration). Our bestprediction of the present-dayDole effect,+20.8 %0,is considerablylower thanthe observedvalue, + 23.5 %0,and we discusspossible causes of this discrepancy. During the past 130 kyr, the Dole effecthas been 0.05 %0lower thanthe present value, on average.The standarddeviation of the Dole effect from the meanhas been only _+0.2 %0,and the Dole effect is nearly unchangedbetween glacial maxima and interglacial periods. The smallvariability in theDole effect suggeststhat relative rates of primaryproduction in theland and marine realms have been relatively constant.Most periodicvariability in the Dole effectis in the precessionband, suggesting that changes in thisglobal biogeochemical term reflects variations in low-latitudeland hydrology and productivity or possiblyvariability in low-latitudeoceanic productivity. Introduction they can potentially be made to do so. Ice core reconstructionsof the concentrationsof bioactivegases in air Our understanding of the response of the biosphere to provide an integratedglobal signal which complementsthe Pleistoceneclimate change is actually quite limited. -

Unilever Time to Lead Us out of the Plastics Crisis © Greenpeace© © Justin© Hofman Greenpeace
Unilever Time to lead us out of the plastics crisis © Greenpeace© © JustinHofman Greenpeace/ 2 Greenpeace Nederland Unilever Time to lead us out of the plastics crisis The problem with plastics Unilever’s plastic footprint and impact Every year, millions of tonnes of plastic waste is polluting our oceans, A 2019 audit of plastic waste (brand audit) by NGO GAIA reveals waterways and communities and impacting our health. Plastic Unilever as the second worst polluter in terms of collected plastic packaging, designed to be used once and thrown away, is one of pollution in the Philippines,7 and it has featured among the top the biggest contributors to the global plastics waste stream.1 The polluters in several other brand audits recently: Unilever was the vast majority of the 8.3 billion tonnes of plastic that has ever been number 2 polluter in a Manila brand audit in 2017, and number produced has been dumped into landfills or has ended up polluting 7 in a global brand audit in 2018, which represented 239 clean- our rivers, oceans, waterways and communities and impacting our ups spanning 42 countries. Therefore Unilever has both a huge health.2 Every year, between 4.8 to 12.7 million tonnes of plastic responsibility for the plastic pollution crisis, and an opportunity to enter our oceans,3 with only nine percent of plastic waste recycled tackle the problem at the source by reducing its use of single-use globally.4 We don’t know exactly how long oil-based plastic will take plastic packaging units. to break down, but once it’s in the environment, it is impossible to clean up; and so the plastic waste crisis continues. -

Dove Packaging Mucell Technology
22 April 2014 ZOTEFOAMS plc ("Zotefoams" or "the Company") Unilever to use Zotefoams’s MuCell® Extrusion technology for its Dove Body Wash bottles in Europe, saving up to 275 tonnes of plastic a year Zotefoams, a world leader in cellular material technology, is pleased to note today’s announcement by Unilever that Unilever’s Dove Body Wash bottles will contain 15% less plastic as a result of a breakthrough packaging technology based on Zotefoams’s MuCell Extrusion microcellular technology. The full text of Unilever’s announcement follows: UNILEVER LAUNCHES BREAKTHROUGH PACKAGING TECHNOLOGY THAT USES 15% LESS PLASTIC Newly developed MuCell® Technology will first feature in Dove Body Wash bottles in Europe, saving up to 275 tonnes of plastic a year London/Rotterdam, 22 April 2014. Dove Body Wash bottles will contain at a minimum 15% less plastic as a result of a newly developed packaging technology launched by Unilever today. Unilever intends to widen the availability of this technology to be used more broadly across the industry. The new technology represents another substantial contribution to the target set out in the Unilever Sustainable Living Plan to halve waste footprint by 2020. The MuCell ® Technology for Extrusion Blow Moulding (EBM) was created in close collaboration with two of Unilever’s global packaging suppliers, Alpla and MuCell Extrusion. It represents a breakthrough in bottle technology: by using gas-injection to create gas bubbles in the middle layer of the bottle wall, it reduces the density of the bottle and the amount of plastic required. The technology will be deployed first in Europe across the Dove Body Wash range, before rolling the technology out. -

YVS STOCK LIST 1St JULY 20
FLAT NUMBER: Type Name Price How Many BAKERY Hovis - Wholemeal £1.60 BAKERY Hovis - Soft White £1.50 BAKERY Pita Bread - white (6) £1.10 BAKERY Granary Bread £1.70 BAKERY Hovis small wholemeal loaf £1.10 BAKERY Pita Bread - wholemeal (6) £1.10 BAKERY DTC - Oven Baked White Baguettes (2) £0.85 BATHROOM & CLEANING Anti-bacterial Handwash (500ml) £1.00 BATHROOM & CLEANING Carex - Anti-bacterial Handwash £1.50 BATHROOM & CLEANING Comfort - Fabric Conditioner (Sunshiny) £1.99 BATHROOM & CLEANING Cushelle - Original (9 roll) £5.49 BATHROOM & CLEANING Toilet DucK Marine 750ml £1.29 BATHROOM & CLEANING Fairy Non Bio Washing Pods x15 £4.49 BATHROOM & CLEANING Domestos - Regular Blue Bleach £1.00 BATHROOM & CLEANING Happy Shopper - Family Tissues £1.00 BATHROOM & CLEANING Imperial Leather - Talcum Powder £1.49 BATHROOM & CLEANING Fairy Washing Up Liquid Orginal 433ml £1.29 BATHROOM & CLEANING Spontex - 2 Washups sponges £0.95 BATHROOM & CLEANING Cif - Lemon (250ml) £1.49 BATHROOM & CLEANING Raid - Fly & Wasp Killer £2.99 BATHROOM & CLEANING Flash Multi Surface Ultra Power Concentrate 400ml £1.49 BATHROOM & CLEANING Flash Spray with bleach £1.91 BATHROOM & CLEANING Bold - 2in1 Washing Powder £2.99 BATHROOM & CLEANING Comfort - Fabric Conditioner (Blue SKies) £1.99 BATHROOM & CLEANING Sponges - Tough Scourers £1.00 BATHROOM & CLEANING Best-one - 3 Sponges £0.59 BATHROOM & CLEANING Dettol surface wipes £6.50 BATHROOM & CLEANING Daz washing liquid £2.99 BATHROOM & CLEANING Persil Washing Powder - Non-Bio £2.99 BATHROOM & CLEANING Andrex - Supreme Quilt -

Compilation of Minimum and Maximum Isotope Ratios of Selected Elements in Naturally Occurring Terrestrial Materials and Reagents by T
U.S. Department of the Interior U.S. Geological Survey Compilation of Minimum and Maximum Isotope Ratios of Selected Elements in Naturally Occurring Terrestrial Materials and Reagents by T. B. Coplen', J. A. Hopple', J. K. Bohlke', H. S. Reiser', S. E. Rieder', H. R. o *< A R 1 fi Krouse , K. J. R. Rosman , T. Ding , R. D. Vocke, Jr., K. M. Revesz, A. Lamberty, P. Taylor, and P. De Bievre6 'U.S. Geological Survey, 431 National Center, Reston, Virginia 20192, USA 2The University of Calgary, Calgary, Alberta T2N 1N4, Canada 3Curtin University of Technology, Perth, Western Australia, 6001, Australia Institute of Mineral Deposits, Chinese Academy of Geological Sciences, Beijing, 100037, China National Institute of Standards and Technology, 100 Bureau Drive, Stop 8391, Gaithersburg, Maryland 20899 "institute for Reference Materials and Measurements, Commission of the European Communities Joint Research Centre, B-2440 Geel, Belgium Water-Resources Investigations Report 01-4222 Reston, Virginia 2002 U.S. Department of the Interior GALE A. NORTON, Secretary U.S. Geological Survey Charles G. Groat, Director The use of trade, brand, or product names in this report is for identification purposes only and does not imply endorsement by the U.S. Government. For additional information contact: Chief, Isotope Fractionation Project U.S. Geological Survey Mail Stop 431 - National Center Reston, Virginia 20192 Copies of this report can be purchased from: U.S. Geological Survey Branch of Information Services Box 25286 Denver, CO 80225-0286 CONTENTS Abstract...................................................... -
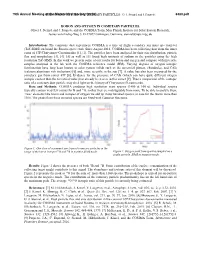
Boron and Oxygen in Cometary Particles: O. J
79th Annual Meeting ofBORON the Meteoritical AND OXYGEN Society IN COMETARY (2016) PARTICLES: O. J. Stenzel and J. Paquette 6380.pdf BORON AND OXYGEN IN COMETARY PARTICLES. Oliver J. Stenzel and J. Paquette and the COSIMA Team, Max Planck Institute for Solar System Research, Justus-von-Liebig-Weg 3, D-37077 Göttingen, Germany, [email protected]. Introduction: The cometary dust experiment COSIMA is a time of flight secondary ion mass spectrometer (ToF-SIMS) on board the Rosetta space craft. Since August 2014, COSIMA has been collecting dust from the inner coma of 67P/Churyumov-Gerasimenko [1], [2]. The particles have been analyzed for their size distribution, particle flux and morphology [3], [4]. [5] as well as [1] found high amounts of sodium in the particles using the high resolution ToF-SIMS. In this work we present some of our results for boron and oxygen and compare with meteorite samples analyzed in the lab with the COSIMA reference model (RM). Varying degrees of oxygen isotopic fractionation have long been known in solar system solids such as the terrestrial planets, chondrules, and CAIs (calcium-aluminum-rich inclusions) [6] and, more recently, in the sun [7]. A value has also been measured for the cometary gas from comet 67P [8]. Evidence for the presence of CAIs (which can have quite different oxygen isotopic content than the terrestrial value) has already been seen in this comet [9]. Thus a comparison of the isotopic ratio of a cometary dust particle may shed light on the history of Churyumov-Gerasimenko. Data and Methods: COSIMA produces high resolution mass spectra (1400 at 100 u). -

Investigation of Nitrogen Cycling Using Stable Nitrogen and Oxygen Isotopes in Narragansett Bay, Ri
University of Rhode Island DigitalCommons@URI Open Access Dissertations 2014 INVESTIGATION OF NITROGEN CYCLING USING STABLE NITROGEN AND OXYGEN ISOTOPES IN NARRAGANSETT BAY, RI Courtney Elizabeth Schmidt University of Rhode Island, [email protected] Follow this and additional works at: https://digitalcommons.uri.edu/oa_diss Recommended Citation Schmidt, Courtney Elizabeth, "INVESTIGATION OF NITROGEN CYCLING USING STABLE NITROGEN AND OXYGEN ISOTOPES IN NARRAGANSETT BAY, RI" (2014). Open Access Dissertations. Paper 209. https://digitalcommons.uri.edu/oa_diss/209 This Dissertation is brought to you for free and open access by DigitalCommons@URI. It has been accepted for inclusion in Open Access Dissertations by an authorized administrator of DigitalCommons@URI. For more information, please contact [email protected]. INVESTIGATION OF NITROGEN CYCLING USING STABLE NITROGEN AND OXYGEN ISOTOPES IN NARRAGANSETT BAY, RI BY COURTNEY ELIZABETH SCHMIDT A DISSERTATION SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY IN OCEANOGRAPHY UNIVERSITY OF RHODE ISLAND 2014 DOCTOR OF PHILOSOPHY DISSERTATION OF COURTNEY ELIZABETH SCHMIDT APPROVED: Thesis Committee: Major Professor Rebecca Robinson Candace Oviatt Arthur Gold Nassar H. Zawia DEAN OF THE GRADUATE SCHOOL UNIVERSITY OF RHODE ISLAND 2014 ABSTRACT Estuaries regulate nitrogen (N) fluxes transported from land to the open ocean through uptake and denitrification. In Narragansett Bay, anthropogenic N loading has increased over the last century with evidence for eutrophication in some regions of Narragansett Bay. Increased concerns over eutrophication prompted upgrades at wastewater treatment facilities (WWTFs) to decrease the amount of nitrogen discharged. The upgrade to tertiary treatment – where bioavailable nitrogen is reduced and removed through denitrification – has occurred at multiple facilities throughout Narragansett Bay’s watershed. -

Unilever to Receive the World Environment Center's 2013 Gold Medal Award for Sustainable Development
Unilever to Receive the World Environment Center’s 2013 Gold Medal Award for Sustainable Development Washington, DC, May 8, 2013 The World Environment Center’s (WEC) 2013 Gold Medal Award for International Corporate Achievement in Sustainable Development will be presented to Unilever CEO Mr. Paul Polman on May 9, 2013 at the 29th Annual WEC Gold Medal Gala in Washington, D.C. Former U.S. Environmental Protection Agency Administrator Lisa P. Jackson will present the award before an audience of global sustainability leaders from business, government, non-governmental organizations, academia and other stakeholders. Unilever is being recognized for its deep and longstanding commitment to advancing environmental sustainability and for its business strategy, the Unilever Sustainable Living Plan, that is applied to its products, governance structure, supply chain and consumers. In accepting the award, Unilever CEO Paul Polman said, “I am delighted that WEC has recognized Unilever with its Gold Medal. The Unilever Sustainable Living Plan is at the heart of our new business model for sustainable, equitable growth and, excitingly, is inspiring people inside and outside the company.” In acknowledging Unilever’s achievements, former Administrator Jackson stated that “Unilever is a prime example of a company that recognizes the need to go beyond improving sustainability performance in its own operations in order to solve global scale problems.” Unilever’s Gold Medal submission was evaluated in a global competition with companies in multiple business sectors by WEC’s independent Gold Medal Jury chaired by Dr. Joel Abrams, professor emeritus at the University of Pittsburgh. The WEC Gold Medal Award is presented annually to a global company that has demonstrated a unique example of sustainability in business practice and is one of the most prestigious forms of recognition of a global company's ongoing commitment to the practice of sustainable development. -

Chemicals Used in the Household
Supplementary Information An Approach for Prioritizing “Down-the-Drain” Chemicals Used in the Household The questionnaire: Please list up to 10 products you most frequently use in the bathroom and kitchen. These should be: • Cleaning products in the kitchen (such as dishwashing liquid, dishwasher powder, fabric conditioner, disinfectant) • Cleaning products in the bathroom (such as bleach, lime scale remover) • Personal care products (such as shampoo, hair conditioner, toothpaste, deodorant, cream soap, soap and body cream) Please identify the product, along with the brand and the exact name of the product. For each of these, please tick how often (daily, weekly or montly) and how much (0–10, 10–100 or >100 mL (or g)) of the product is used each day/week/month. Two examples are given below. Frequency Quantity Used Each Day/Week/Month Product Brand 0–10 mL 1–100 mL >100 mL Daily Weekly Monthly (or g) (or g) (or g) Dishwashing liquid FAIRY clean and fresh (apple and orchard) Toothpaste SENSODYNE Daily care 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. Int. J. Environ. Res. Public Health 2015, 12 S2 Table S1. The 26 different hand wash gels as reported by the respondents who used these products, in order of decreasing average use. Frequency Estimate of Use (mL) Average Use Brand Full Description Users Daily 0–10 10–100 >100 L·per-1·yr-1 Palmolive Milk and Honey 4 4 4 0.83 Simple Kind to Skin (Antibacterial) 3 3 1 2 0.62 Cien Water Lily and Lotus 2 2 2 0.42 Dove Beauty Cream Wash 2 2 2 0.42 (Cussons) Protect Plus Carex 2 2 1 1 0.23 (Antibacterial) Dettol -

Press Release
Classification: General Business Use PRESS RELEASE Sittard, The Netherlands, 24th January 2019 SABIC AND CUSTOMERS LAUNCH CERTIFIED CIRCULAR POLYMERS FROM MIXED PLASTIC WASTE SABIC and customers Unilever, Vinventions and Walki Group will introduce ISCC certified circular polymers in 2019 during a market foundation stage. SABIC’s certified circular polymers will be produced in The Netherlands from a recycled plastic waste feedstock developed by PLASTIC ENERGY and offer a drop-in alternative for customers looking at meeting the needs of various challenging applications. The initiative to upcycle mixed plastic waste back to the original polymer supports SABIC’s and its feedstock supplier and customers commitment to providing innovative solutions for a circular economy. SABIC, a global leader in the chemical industry, has announced together with its customers Unilever, Vinventions and Walki Group, the launch of certified circular polymers to be manufactured by SABIC and planned to be used by its customers for packaging solutions for a variety of consumer products that will be introduced into the market in 2019. The certified circular polymers will be produced from a feedstock known as TACOIL – a patented product from UK-based PLASTIC ENERGY Ltd - from the recycling of low quality, mixed plastic waste otherwise destined for incineration or landfill. SABIC will process this feedstock on its production site at Geleen in The Netherlands. The finished certified circular polymers will then be supplied to the three key customers to use in their development of pioneering, high quality and safe consumer packaging for food, beverage, personal and home care products. The market foundation stage is an important step of a project recently announced by SABIC and Plastic Energy to build first commercial plants in the Netherlands to manufacture and process the feedstock.