Novel Approaches to the Analysis of Steroid Estrogens in River Sediments
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-
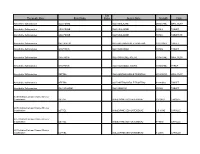
Therapeutic Class Brand Name P a Status Generic
P A Therapeutic Class Brand Name Status Generic Name Strength Form Absorbable Sulfonamides AZULFIDINE SULFASALAZINE 250MG/5ML ORAL SUSP Absorbable Sulfonamides AZULFIDINE SULFASALAZINE 500MG TABLET Absorbable Sulfonamides AZULFIDINE SULFASALAZINE 500MG TABLET DR Absorbable Sulfonamides BACTRIM DS SULFAMETHOXAZOLE/TRIMETHO 800-160MG TABLET Absorbable Sulfonamides GANTRISIN SULFISOXAZOLE 500MG TABLET Absorbable Sulfonamides GANTRISIN SULFISOXAZOLE ACETYL 500MG/5ML ORAL SUSP Absorbable Sulfonamides GANTRISIN SULFISOXAZOLE ACETYL 500MG/5ML SYRUP Absorbable Sulfonamides SEPTRA SULFAMETHOXAZOLE/TRIMETHO 200-40MG/5 ORAL SUSP Absorbable Sulfonamides SEPTRA SULFAMETHOXAZOLE/TRIMETHO 400-80MG TABLET Absorbable Sulfonamides SULFADIAZINE SULFADIAZINE 500MG TABLET ACE Inhibitor/Calcium Channel Blocker Combination LOTREL AMLODIPINE BESYLATE/BENAZ 10-20MG CAPSULE ACE Inhibitor/Calcium Channel Blocker Combination LOTREL AMLODIPINE BESYLATE/BENAZ 2.5-10MG CAPSULE ACE Inhibitor/Calcium Channel Blocker Combination LOTREL AMLODIPINE BESYLATE/BENAZ 5-10MG CAPSULE ACE Inhibitor/Calcium Channel Blocker Combination LOTREL AMLODIPINE BESYLATE/BENAZ 5-20MG CAPSULE P A Therapeutic Class Brand Name Status Generic Name Strength Form ACE Inhibitor/Calcium Channel Blocker Combination LOTREL AMLODIPINE BESYLATE/BENAZ 5-40MG CAPSULE ACE Inhibitor/Calcium Channel Blocker Combination LOTREL AMLODIPINE BESYLATE/BENAZ 10-40MG CAPSULE Acne Agents, Systemic ACCUTANE ISOTRETINOIN 10MG CAPSULE Acne Agents, Systemic ACCUTANE ISOTRETINOIN 20MG CAPSULE Acne Agents, Systemic ACCUTANE -

Postmenopausal Pharmacotherapy Newsletter
POSTMENOPAUSAL PHARMACOTHERAPY September, 1999 As Canada's baby boomers age, more and more women will face the option of Hormone Replacement Therapy (HRT). The HIGHLIGHTS decision can be a difficult one given the conflicting pros and cons. M This RxFiles examines the role and use of HRT, as well as newer Long term HRT carries several major benefits but also risks SERMS and bisphosphonates in post-menopausal (PM) patients. which should be evaluated on an individual and ongoing basis MContinuous ERT is appropriate for women without a uterus HRT MWomen with a uterus should receive progestagen (at least 12 HRT is indicated for the treatment of PM symptoms such as days per month or continuous low-dose) as part of their HRT vasomotor disturbances and urogenital atrophy, and is considered MLow-dose ERT (CEE 0.3mg) + Ca++ appears to prevent PMO primary therapy for prevention and treatment of postmenopausal MBisphosphinates (e.g. alendronate, etidronate) and raloxifene are osteoporosis (PMO).1 Contraindications are reviewed in Table 2. alternatives to HRT in treating and preventing PMO Although HRT is contraindicated in women with active breast or M"Natural" HRT regimens can be compounded but data is lacking uterine cancer, note that a prior or positive family history of these does not necessarily preclude women from receiving HRT.1 Comparative Safety: Because of differences between products, some side effects may be alleviated by switching from one product Estrogen Replacement Therapy (ERT) 2 to another, particularly from equine to plant sources or from oral to Naturally secreted estrogens include: topical (see Table 3 - Side Effects & Their Management). -

Title 16. Crimes and Offenses Chapter 13. Controlled Substances Article 1
TITLE 16. CRIMES AND OFFENSES CHAPTER 13. CONTROLLED SUBSTANCES ARTICLE 1. GENERAL PROVISIONS § 16-13-1. Drug related objects (a) As used in this Code section, the term: (1) "Controlled substance" shall have the same meaning as defined in Article 2 of this chapter, relating to controlled substances. For the purposes of this Code section, the term "controlled substance" shall include marijuana as defined by paragraph (16) of Code Section 16-13-21. (2) "Dangerous drug" shall have the same meaning as defined in Article 3 of this chapter, relating to dangerous drugs. (3) "Drug related object" means any machine, instrument, tool, equipment, contrivance, or device which an average person would reasonably conclude is intended to be used for one or more of the following purposes: (A) To introduce into the human body any dangerous drug or controlled substance under circumstances in violation of the laws of this state; (B) To enhance the effect on the human body of any dangerous drug or controlled substance under circumstances in violation of the laws of this state; (C) To conceal any quantity of any dangerous drug or controlled substance under circumstances in violation of the laws of this state; or (D) To test the strength, effectiveness, or purity of any dangerous drug or controlled substance under circumstances in violation of the laws of this state. (4) "Knowingly" means having general knowledge that a machine, instrument, tool, item of equipment, contrivance, or device is a drug related object or having reasonable grounds to believe that any such object is or may, to an average person, appear to be a drug related object. -

Hormone Replacement Therapy and Osteoporosis
This report may be used, in whole or in part, as the basis for development of clinical practice guidelines and other quality enhancement tools, or a basis for reimbursement and coverage policies. AHRQ or U.S. Department of Health and Human Services endorsement of such derivative products may not be stated or implied. AHRQ is the lead Federal agency charged with supporting research designed to improve the quality of health care, reduce its cost, address patient safety and medical errors, and broaden access to essential services. AHRQ sponsors and conducts research that provides evidence-based information on health care outcomes; quality; and cost, use, and access. The information helps health care decisionmakers— patients and clinicians, health system leaders, and policymakers—make more informed decisions and improve the quality of health care services. Systematic Evidence Review Number 12 Hormone Replacement Therapy and Osteoporosis Prepared for: Agency for Healthcare Research and Quality U.S. Department of Health and Human Services 2101 East Jefferson Street Rockville, MD 20852 http://www.ahrq.gov Contract No. 290-97-0018 Task Order No. 2 Technical Support of the U.S. Preventive Services Task Force Prepared by: Oregon Health Sciences University Evidence-based Practice Center, Portland, Oregon Heidi D. Nelson, MD, MPH August 2002 Preface The Agency for Healthcare Research and Quality (AHRQ) sponsors the development of Systematic Evidence Reviews (SERs) through its Evidence-based Practice Program. With guidance from the third U.S. Preventive Services Task Force∗ (USPSTF) and input from Federal partners and primary care specialty societies, two Evidence-based Practice Centers—one at the Oregon Health Sciences University and the other at Research Triangle Institute-University of North Carolina—systematically review the evidence of the effectiveness of a wide range of clinical preventive services, including screening, counseling, immunizations, and chemoprevention, in the primary care setting. -

Steroidal Estrogens
FINAL Report on Carcinogens Background Document for Steroidal Estrogens December 13 - 14, 2000 Meeting of the NTP Board of Scientific Counselors Report on Carcinogens Subcommittee Prepared for the: U.S. Department of Health and Human Services Public Health Service National Toxicology Program Research Triangle Park, NC 27709 Prepared by: Technology Planning and Management Corporation Canterbury Hall, Suite 310 4815 Emperor Blvd Durham, NC 27703 Contract Number N01-ES-85421 Dec. 2000 RoC Background Document for Steroidal Estrogens Do not quote or cite Criteria for Listing Agents, Substances or Mixtures in the Report on Carcinogens U.S. Department of Health and Human Services National Toxicology Program Known to be Human Carcinogens: There is sufficient evidence of carcinogenicity from studies in humans, which indicates a causal relationship between exposure to the agent, substance or mixture and human cancer. Reasonably Anticipated to be Human Carcinogens: There is limited evidence of carcinogenicity from studies in humans which indicates that causal interpretation is credible but that alternative explanations such as chance, bias or confounding factors could not adequately be excluded; or There is sufficient evidence of carcinogenicity from studies in experimental animals which indicates there is an increased incidence of malignant and/or a combination of malignant and benign tumors: (1) in multiple species, or at multiple tissue sites, or (2) by multiple routes of exposure, or (3) to an unusual degree with regard to incidence, site or type of tumor or age at onset; or There is less than sufficient evidence of carcinogenicity in humans or laboratory animals, however; the agent, substance or mixture belongs to a well defined, structurally-related class of substances whose members are listed in a previous Report on Carcinogens as either a known to be human carcinogen, or reasonably anticipated to be human carcinogen or there is convincing relevant information that the agent acts through mechanisms indicating it would likely cause cancer in humans. -

The Rabbits Are Prepared ..." - the Development of Ethinylestradiol and Ethinyltestosterone Frobenius W J
Journal für Reproduktionsmedizin und Endokrinologie – Journal of Reproductive Medicine and Endocrinology – Andrologie • Embryologie & Biologie • Endokrinologie • Ethik & Recht • Genetik Gynäkologie • Kontrazeption • Psychosomatik • Reproduktionsmedizin • Urologie "The Rabbits are Prepared ..." - The Development of Ethinylestradiol and Ethinyltestosterone Frobenius W J. Reproduktionsmed. Endokrinol 2011; 8 (Sonderheft 1), 32-57 www.kup.at/repromedizin Online-Datenbank mit Autoren- und Stichwortsuche Offizielles Organ: AGRBM, BRZ, DVR, DGA, DGGEF, DGRM, D·I·R, EFA, OEGRM, SRBM/DGE Indexed in EMBASE/Excerpta Medica/Scopus Krause & Pachernegg GmbH, Verlag für Medizin und Wirtschaft, A-3003 Gablitz FERRING-Symposium digitaler DVR 2021 Mission possible – personalisierte Medizin in der Reproduktionsmedizin Was kann die personalisierte Kinderwunschbehandlung in der Praxis leisten? Freuen Sie sich auf eine spannende Diskussion auf Basis aktueller Studiendaten. SAVE THE DATE 02.10.2021 Programm 12.30 – 13.20Uhr Chair: Prof. Dr. med. univ. Georg Griesinger, M.Sc. 12:30 Begrüßung Prof. Dr. med. univ. Georg Griesinger, M.Sc. & Dr. Thomas Leiers 12:35 Sind Sie bereit für die nächste Generation rFSH? Im Gespräch Prof. Dr. med. univ. Georg Griesinger, Dr. med. David S. Sauer, Dr. med. Annette Bachmann 13:05 Die smarte Erfolgsformel: Value Based Healthcare Bianca Koens 13:15 Verleihung Frederik Paulsen Preis 2021 Wir freuen uns auf Sie! Development of Ethinylestradiol and Ethinyltestosterone “The Rabbits are Prepared …” – The Development of Ethinylestradiol and Ethinyltestosterone W. Frobenius In an exciting scientific neck-and-neck race, European and American scientists in the late 1920s and early 1930s isolated the ovarian, placental, and testicular hormones. At the same time the constitution of the human sex steroids was elucidated. However, it soon emerged that with oral administration the therapeutic value of the natural substances was extremely limited. -
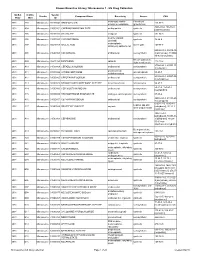
Known Bioactive Library: Microsource 1 - US Drug Collection
Known Bioactive Library: Microsource 1 - US Drug Collection ICCB-L ICCB-L Vendor Vendor Compound Name Bioactivity Source CAS Plate Well ID antifungal, inhibits Penicillium 2091 A03 Microsource 00200046 GRISEOFULVIN 126-07-8 mitosis in metaphase griseofulvum 3505-38-2, 486-16-8 2091 A04 Microsource 01500161 CARBINOXAMINE MALEATE antihistaminic synthetic [carbinoxamine] 2091 A05 Microsource 00200331 SALSALATE analgesic synthetic 552-94-3 muscle relaxant 2091 A06 Microsource 01500162 CARISOPRODOL synthetic 78-44-4 (skeletal) antineoplastic, 2091 A07 Microsource 00210369 GALLIC ACID insect galls 149-91-7 astringent, antibacterial 66592-87-8, 50370-12- 2091 A08 Microsource 01500163 CEFADROXIL antibacterial semisynthetic 2 [anhydrous], 119922- 89-9 [hemihydrate] Rheum palmatum, 2091 A09 Microsource 00211468 DANTHRON cathartic 117-10-2 Xyris semifuscata 27164-46-1, 25953-19- 2091 A10 Microsource 01500164 CEFAZOLIN SODIUM antibacterial semisynthetic 9 [cefazolin] glucocorticoid, 2091 A11 Microsource 00300024 HYDROCORTISONE adrenal glands 50-23-7 antiinflammatory 64485-93-4, 63527-52- 2091 A12 Microsource 01500165 CEFOTAXIME SODIUM antibacterial semisynthetic 6 [cefotaxime] 2091 A13 Microsource 00300029 DESOXYCORTICOSTERONE ACETATE mineralocorticoid adrenocortex 56-47-3 58-71-9, 153-61-7 2091 A14 Microsource 01500166 CEPHALOTHIN SODIUM antibacterial semisynthetic [cephalothin] 2091 A15 Microsource 00300034 TESTOSTERONE PROPIONATE androgen, antineoplastic semisynthetic 57-85-2 24356-60-3, 21593-23- 2091 A16 Microsource 01500167 CEPHAPIRIN SODIUM -

CYTOTOXIC and NON-CYTOTOXIC HAZARDOUS MEDICATIONS
CYTOTOXIC and NON-CYTOTOXIC HAZARDOUS MEDICATIONS1 CYTOTOXIC HAZARDOUS MEDICATIONS NON-CYTOTOXIC HAZARDOUS MEDICATIONS Altretamine IDArubicin Acitretin Iloprost Amsacrine Ifosfamide Aldesleukin Imatinib 3 Arsenic Irinotecan Alitretinoin Interferons Asparaginase Lenalidomide Anastrazole 3 ISOtretinoin azaCITIDine Lomustine Ambrisentan Leflunomide 3 azaTHIOprine 3 Mechlorethamine Bacillus Calmette Guerin 2 Letrozole 3 Bleomycin Melphalan (bladder instillation only) Leuprolide Bortezomib Mercaptopurine Bexarotene Megestrol 3 Busulfan 3 Methotrexate Bicalutamide 3 Methacholine Capecitabine 3 MitoMYcin Bosentan MethylTESTOSTERone CARBOplatin MitoXANtrone Buserelin Mifepristone Carmustine Nelarabine Cetrorelix Misoprostol Chlorambucil Oxaliplatin Choriogonadotropin alfa Mitotane CISplatin PACLitaxel Cidofovir Mycophenolate mofetil Cladribine Pegasparaginase ClomiPHENE Nafarelin Clofarabine PEMEtrexed Colchicine 3 Nilutamide 3 Cyclophosphamide Pentostatin cycloSPORINE Oxandrolone 3 Cytarabine Procarbazine3 Cyproterone Pentamidine (Aerosol only) Dacarbazine Raltitrexed Dienestrol Podofilox DACTINomycin SORAfenib Dinoprostone 3 Podophyllum resin DAUNOrubicin Streptozocin Dutasteride Raloxifene 3 Dexrazoxane SUNItinib Erlotinib 3 Ribavirin DOCEtaxel Temozolomide Everolimus Sirolimus DOXOrubicin Temsirolimus Exemestane 3 Tacrolimus Epirubicin Teniposide Finasteride 3 Tamoxifen 3 Estramustine Thalidomide Fluoxymesterone 3 Testosterone Etoposide Thioguanine Flutamide 3 Tretinoin Floxuridine Thiotepa Foscarnet Trifluridine Flucytosine Topotecan Fulvestrant -

Bioidentical Hormones: an Evidence-Based Review for Primary Care Providers
REVIEW Bioidentical Hormones: An Evidence-Based Review for Primary Care Providers Eileen Conaway, DO Context: Since 2002, when the US Food and Drug Adminis - combination with estrogen for the management of vasomotor tration (FDA) placed a black box warning on women’s hor - symptoms. Dehydroepiandrosterone is not FDA approved, mone replacement products, women and their providers but small-scale studies indicate it may improve bone min - have been struggling with whether to proceed with hormone eral density. Data are conflicting about efficacy in improving replacement therapy. Out of the controversy has grown a sexual dysfunction. There is an abundance of misleading popular movement promoting the use of bioidentical hor - information available in the media and on the Internet for mones. Many providers are still unsure if they want to rec - our patients. Compounded bioidenticals and salivary hor - ommend these products and, if so, how to use them appro - mone testing are unnecessary, are not standardized, and priately. should be avoided. Objective: To inform primary care providers (eg, physicians, Conclusion: Bioidentical hormones that are approved by the physician assistants, nurse practitioners) about current data on FDA may be preferred over standard hormone replacement the safety and efficacy of bioidentical hormone replacement because of their physiologic benefits and safety profile. therapy and to provide a context for patient perceptions. J Am Osteopath Assoc . 2011;111(3):153-164 Methods: Literature published between 1999 and 2009 was reviewed through MD Consult’s Medline and Ovid search n the aftermath of the unexpected adverse results of the engines. A Google search of popular media was also per - IWomen’s Health Initiative (WHI) trial 1 in 2002, women formed using the same terms. -

Studies on the Chemical and Biological Properties of Coumestrol and Related Compounds
C/^% Studies on the Chemical and Biological Properties of Coumestrol and Related Compounds TechnicaLBuUetin No. 1408 îsJ ■■-1 r Agricultural Research Service UNITED STATES DEPARTMENT OF AGRICULTURE 6 Studies on the Chemical and Biological Properties of Coumestrol and Related Compounds Technical Bulletin No. 1408 Agricultural Research Service UNITED STATES DEPARTMENT OF AGRICULTURE Contents Page Page Introduction 1 Synthesis of coumestans—con. Analytical procedures 3 Synthesis from previously Biological procedures 6 formed coumestans 32 Chromatographie procedures 7 Physical properties of coumestans Qualitative procedure 8 Ultraviolet absorption spectral Quantitative procedure 9 measurements 32 Other Chromatographie pro- Proton magnetic resonance cedures 10 spectral measurements 35 Isolation 14 Mass spectral measurements 37 Isolation of coumestrol from Chromatography 41 Ladino clover 15 Coumestrol in plants 43 Large-scale processing of al- Variations as affected by cut- falfa for coumestrol 16 tings and stages of growth __ 43 Isolation of compounds closely- Variations as aifected by loca- related to coumestrol 17 tion and climatic conditions __ 46 Characterization of coumestans __ 18 Variations as aifected by varietal Coumestrol 20 and genetic differences 47 Trifoliol 20 Variations as related to disease. 48 Medicagol 24 Mechanism of biosynthesis 53 4'-0-methylcoumestrol 24 Utilization of coumestrol by the 3'-Methoxycoumestrol 24 animal 55 Lucernol 25 Metabolism 58 Sativol 25 Estrogenic potency 59 11, 12-Dimethoxy-7-hydroxy- Antifertility -

Drugs for the Treatment of Menopausal Symptoms
Review Drugs for the treatment of menopausal symptoms † Susan R Davis & Fiona Jane Monash University, Alfred Hospital, Central Clinical School, Women’s Health Program, 1. Introduction Commercial Road, Prahran, Victoria, Australia 2. Which menopausal symptoms Importance of the field: Over the last decade, the management of the meno- merit treatment? pause has attracted extensive public and professional debate and has become 3. Pharmacotherapy for one of the most controversial areas in clinical practice. menopausal symptoms Areas covered in this review: This review provides an overview of the field, 4. New frontiers primarily from a clinical practice perspective. However, as we have incorpo- 5. Expert opinion rated in this ‘big-picture’ snapshot of the field both conventional and comple- mentary approaches to managing the menopause, it is not an exhaustive review of the literature. What the reader will gain: By reviewing menopausal management from the perspective of practicing clinicians, we hope readers will gain insight into decision making processes appropriate for dealing with symptomatic women. Take home message: Although most women do not require pharmacother- apy for menopausal symptoms, many are severely affected by estrogen defi- ciency at and beyond menopause and, for such women, hormone therapy is important if they are to retain an acceptable quality of life. This article consid- ers the drug treatment of the symptomatic postmenopausal woman and the safety issues related to these medications. Keywords: estrogen, HRT, menopause, progestogen Expert Opin. Pharmacother. (2010) 11(8):1329-1341 For personal use only. 1. Introduction Menopause is not only associated with symptoms that range from bothersome through to extremely distressing, it is also accompanied by hormonal changes that impact adversely on several non-reproductive systems. -

PRAC Recommendations on Signals Adopted at the 13-16 January 2020 PRAC En
10 February 20201 EMA/PRAC/8637/2020 Pharmacovigilance Risk Assessment Committee (PRAC) PRAC recommendations on signals Adopted at the 13-16 January 2020 PRAC meeting This document provides an overview of the recommendations adopted by the Pharmacovigilance Risk Assessment Committee (PRAC) on the signals discussed during the meeting of 13-16 January 2020 (including the signal European Pharmacovigilance Issues Tracking Tool [EPITT]2 reference numbers). PRAC recommendations to provide supplementary information are directly actionable by the concerned marketing authorisation holders (MAHs). PRAC recommendations for regulatory action (e.g. amendment of the product information) are submitted to the Committee for Medicinal Products for Human Use (CHMP) for endorsement when the signal concerns Centrally Authorised Products (CAPs), and to the Co-ordination Group for Mutual Recognition and Decentralised Procedures – Human (CMDh) for information in the case of Nationally Authorised Products (NAPs). Thereafter, MAHs are expected to take action according to the PRAC recommendations. When appropriate, the PRAC may also recommend the conduct of additional analyses by the Agency or Member States. MAHs are reminded that in line with Article 16(3) of Regulation No (EU) 726/2004 and Article 23(3) of Directive 2001/83/EC, they shall ensure that their product information is kept up to date with the current scientific knowledge including the conclusions of the assessment and recommendations published on the European Medicines Agency (EMA) website (currently acting as the EU medicines webportal). For CAPs, at the time of publication, PRAC recommendations for update of product information have been agreed by the CHMP at their plenary meeting (27-30 January 2020) and corresponding variations will be assessed by the CHMP.