Detecting Disease Genes Based on Semi-Supervised Learning and Protein–Protein
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Anti-ACTN2 Monoclonal Antibody, Clone FQ3630Z (DCABH-9438) This Product Is for Research Use Only and Is Not Intended for Diagnostic Use
Anti-ACTN2 monoclonal antibody, clone FQ3630Z (DCABH-9438) This product is for research use only and is not intended for diagnostic use. PRODUCT INFORMATION Product Overview Rabbit monoclonal to Sarcomeric Alpha Actinin Antigen Description F-actin cross-linking protein which is thought to anchor actin to a variety of intracellular structures. This is a bundling protein. Immunogen A synthetic peptide (the amino acid sequence is considered to be commercially sensitive) corresponding to residues on the N-terminus of human Sarcomeric Alpha Actinin Isotype IgG Source/Host Rabbit Species Reactivity Mouse, Rat, Human Clone FQ3630Z Purity Tissue culture supernatant Conjugate Unconjugated Applications WB, IP, IHC-P Positive Control Human skeletal muscle, rat heart and human heart lysates and human muscle tissue. Format Liquid Size 100 μl Buffer pH: 7.40; Preservative: 0.01% Sodium azide; Constituents: 50% Glycerol, 0.05% BSA Preservative 0.01% Sodium Azide Storage Store at -20°C. Stable for 12 months at -20°C GENE INFORMATION Gene Name ACTN2 actinin, alpha 2 [ Homo sapiens ] Official Symbol ACTN2 45-1 Ramsey Road, Shirley, NY 11967, USA Email: [email protected] Tel: 1-631-624-4882 Fax: 1-631-938-8221 1 © Creative Diagnostics All Rights Reserved Synonyms ACTN2; actinin, alpha 2; alpha-actinin-2; F-actin cross-linking protein; alpha-actinin skeletal muscle; CMD1AA; Entrez Gene ID 88 Protein Refseq NP_001094 UniProt ID P35609 Chromosome Location 1q42-q43 Pathway Activation of NMDA receptor upon glutamate binding and postsynaptic events, -

Genetic Cardiomyopathies: the Lesson Learned from Hipscs
Journal of Clinical Medicine Review Genetic Cardiomyopathies: The Lesson Learned from hiPSCs Ilaria My 1,2 and Elisa Di Pasquale 2,3,* 1 Department of Biomedical Sciences, Humanitas University, Pieve Emanuele, 20090 Milan, Italy; [email protected] 2 Humanitas Clinical and Research Center—IRCCS, Rozzano, 20089 Milan, Italy 3 Institute of Genetic and Biomedical Research (IRGB)—UOS of Milan, National Research Council (CNR), 20138 Milan, Italy * Correspondence: [email protected] Abstract: Genetic cardiomyopathies represent a wide spectrum of inherited diseases and constitute an important cause of morbidity and mortality among young people, which can manifest with heart failure, arrhythmias, and/or sudden cardiac death. Multiple underlying genetic variants and molecular pathways have been discovered in recent years; however, assessing the pathogenicity of new variants often needs in-depth characterization in order to ascertain a causal role in the disease. The application of human induced pluripotent stem cells has greatly helped to advance our knowledge in this field and enabled to obtain numerous in vitro patient-specific cellular models useful to study the underlying molecular mechanisms and test new therapeutic strategies. A milestone in the research of genetically determined heart disease was the introduction of genomic technologies that provided unparalleled opportunities to explore the genetic architecture of cardiomyopathies, thanks to the generation of isogenic pairs. The aim of this review is to provide an overview of the main research that helped elucidate the pathophysiology of the most common genetic cardiomyopathies: hypertrophic, dilated, arrhythmogenic, and left ventricular noncompaction cardiomyopathies. A special focus is provided on the application of gene-editing techniques in understanding key disease characteristics and on the therapeutic approaches that have been tested. -

Final Draft Masterthesis Gabbin
MASTER THESIS Thesis submitted in partial fulfillment of the requirements for the degree of Master of Science in Engineering at the University of Applied Sciences Technikum Wien Degree Program in Tissue Engineering and Regenerative Medicine Modeling of a Novel Filamin-C Mutation in Restrictive Cardiomyopathy using hiPSC- derived Cardiomyocytes By: Beatrice Gabbin, BSc Student Number: 1810692024 Supervisor 1: Dr. William Pu, MD Dr. Maksymilian Prondzynski, PhD Supervisor 2: Dorota Szwarc, MSc Boston, September 29, 2020 Declaration of Authenticity “As author and creator of this work to hand, I confirm with my signature knowledge of the relevant copyright regulations governed by higher education acts (see Urheberrechtsgesetz/ Austrian copyright law as amended as well as the Statute on Studies Act Provisions / Examination Regulations of the UAS Technikum Wien as amended). I hereby declare that I completed the present work independently and that any ideas, whether written by others or by myself, have been fully sourced and referenced. I am aware of any consequences I may face on the part of the degree program director if there should be evidence of missing autonomy and independence or evidence of any intent to fraudulently achieve a pass mark for this work (see Statute on Studies Act Provisions / Examination Regulations of the UAS Technikum Wien as amended). I further declare that up to this date I have not published the work to hand nor have I presented it to another examination board in the same or similar form. I affirm that the version submitted matches the version in the upload tool.” Boston, September 29, 2020 Place, Date Signature Abstract The purpose of this project was to establish an in vitro model for a novel FLNC missense mutation (c.7416_7418delGAA/p.Glu2472_Asn2473delAsp), which was identified at the Boston Children’s Hospital in a patient diagnosed with restrictive cardiomyopathy (RCM). -

Induction of Therapeutic Tissue Tolerance Foxp3 Expression Is
Downloaded from http://www.jimmunol.org/ by guest on October 2, 2021 is online at: average * The Journal of Immunology , 13 of which you can access for free at: 2012; 189:3947-3956; Prepublished online 17 from submission to initial decision 4 weeks from acceptance to publication September 2012; doi: 10.4049/jimmunol.1200449 http://www.jimmunol.org/content/189/8/3947 Foxp3 Expression Is Required for the Induction of Therapeutic Tissue Tolerance Frederico S. Regateiro, Ye Chen, Adrian R. Kendal, Robert Hilbrands, Elizabeth Adams, Stephen P. Cobbold, Jianbo Ma, Kristian G. Andersen, Alexander G. Betz, Mindy Zhang, Shruti Madhiwalla, Bruce Roberts, Herman Waldmann, Kathleen F. Nolan and Duncan Howie J Immunol cites 35 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription http://www.jimmunol.org/content/suppl/2012/09/17/jimmunol.120044 9.DC1 This article http://www.jimmunol.org/content/189/8/3947.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2012 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of October 2, 2021. -
![Viewed in [38])](https://docslib.b-cdn.net/cover/8742/viewed-in-38-3158742.webp)
Viewed in [38])
Molecular Cancer BioMed Central Research Open Access The parafibromin tumor suppressor protein interacts with actin-binding proteins actinin-2 and actinin-3 Sunita K Agarwal*, William F Simonds and Stephen J Marx Address: National Institute of Diabetes and Digestive and Kidney Diseases, NIH, Bethesda, Maryland, USA Email: Sunita K Agarwal* - [email protected]; William F Simonds - [email protected]; Stephen J Marx - [email protected] * Corresponding author Published: 7 August 2008 Received: 24 April 2008 Accepted: 7 August 2008 Molecular Cancer 2008, 7:65 doi:10.1186/1476-4598-7-65 This article is available from: http://www.molecular-cancer.com/content/7/1/65 © 2008 Agarwal et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Germline and somatic inactivating mutations in the HRPT2 gene occur in the inherited hyperparathyroidism-jaw tumor syndrome, in some cases of parathyroid cancer and in some cases of familial hyperparathyroidism. HRPT2 encodes parafibromin. To identify parafibromin interacting proteins we used the yeast two-hybrid system for screening a heart cDNA library with parafibromin as the bait. Results: Fourteen parafibromin interaction positive preys representing 10 independent clones encoding actinin-2 were isolated. Parafibromin interacted with muscle alpha-actinins (actinin-2 and actinin-3), but not with non-muscle alpha-actinins (actinin-1 and actinin-4). The parafibromin-actinin interaction was verified by yeast two-hybrid, GST pull-down, and co-immunoprecipitation. -

Qt5q82m1wm Nosplash 4742E
Copyright 2014 by Hengameh Zahed ii Acknowledgements My PhD experience, like that of many, has been a journey of self discovery as well as a scientific one. I have been truly lucky to benefit from the help of many on this journey. First and foremost, I dedicate this thesis to my parents, Iran Sabet and Jalil Zahed, who gave up all material possessions, family ties, and social roots to move my siblings and me to the United States so we could pursue the best education and opportunities. Their dreams were large, their bravery unparalleled, and their dedication undeniable. I can never repay them for their sacrifices. I am also grateful to my parents for giving me two partners in crime to go through life with: my hard- working, brilliant, loving siblings, whose support I have relied so heavily on. My sister, Chakameh Zahed, is a perfect embodiment of a resilient mind meeting the most generous of souls. Thank you for being the older sister I could look up to and for helping me find the will to push through hard times. I learned to not shy away from a challenge watching you boldly become a talented and successful “EECS geek” and excel in a heavily male-dominated field. My brother, Hazhir Zahed, has been the voice of calm and reason throughout my life. I am impressed by your confidence and articulateness and have sought your wisdom time and again. From you I’ve learned that success is the end product of gracefully getting up every time you fall. Your drive and passion in everything you do is inspiring. -

Program in Human Neutrophils Fails To
Downloaded from http://www.jimmunol.org/ by guest on September 25, 2021 is online at: average * The Journal of Immunology Anaplasma phagocytophilum , 20 of which you can access for free at: 2005; 174:6364-6372; ; from submission to initial decision 4 weeks from acceptance to publication J Immunol doi: 10.4049/jimmunol.174.10.6364 http://www.jimmunol.org/content/174/10/6364 Insights into Pathogen Immune Evasion Mechanisms: Fails to Induce an Apoptosis Differentiation Program in Human Neutrophils Dori L. Borjesson, Scott D. Kobayashi, Adeline R. Whitney, Jovanka M. Voyich, Cynthia M. Argue and Frank R. DeLeo cites 28 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2005/05/03/174.10.6364.DC1 This article http://www.jimmunol.org/content/174/10/6364.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* • Why • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2005 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 25, 2021. The Journal of Immunology Insights into Pathogen Immune Evasion Mechanisms: Anaplasma phagocytophilum Fails to Induce an Apoptosis Differentiation Program in Human Neutrophils1 Dori L. -

Advanced Evolution of Pathogenesis Concepts in Cardiomyopathies
Journal of Clinical Medicine Review Advanced Evolution of Pathogenesis Concepts in Cardiomyopathies 1, 2,3, 2,3 4 Chia-Jung Li y , Chien-Sheng Chen y, Giou-Teng Yiang , Andy Po-Yi Tsai , Wan-Ting Liao 5,6,* and Meng-Yu Wu 2,3,* 1 Department of Obstetrics and Gynecology, Kaohsiung Veterans General Hospital, Kaohsiung 813, Taiwan; [email protected] 2 Department of Emergency Medicine, Taipei Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation, New Taipei 231, Taiwan; [email protected] (C.-S.C.); [email protected] (G.-T.Y.) 3 Department of Emergency Medicine, School of Medicine, Tzu Chi University, Hualien 970, Taiwan 4 Department of Medical Research, Buddhist Tzu Chi General Hospital, Hualien 970, Taiwan; [email protected] 5 Institute of Medicine, Chung Shan Medical University, Taichung 402, Taiwan 6 Chinese Medicine Department, Show Chwan Memorial Hospital, Changhua 500, Taiwan * Correspondence: [email protected] (W.-T.L.); [email protected] (M.-Y.W.); Tel.: +886-4-725-6166 (W.-T.L.); +886-2-6628-42752 (M.-Y.W.) These authors contributed equally to this work. y Received: 7 March 2019; Accepted: 12 April 2019; Published: 16 April 2019 Abstract: Cardiomyopathy is a group of heterogeneous cardiac diseases that impair systolic and diastolic function, and can induce chronic heart failure and sudden cardiac death. Cardiomyopathy is prevalent in the general population, with high morbidity and mortality rates, and contributes to nearly 20% of sudden cardiac deaths in younger individuals. Genetic mutations associated with cardiomyopathy play a key role in disease formation, especially the mutation of sarcomere encoding genes and ATP kinase genes, such as titin, lamin A/C, myosin heavy chain 7, and troponin T1. -

ALLEN Human Brain Atlas
ALLEN Human Brain Atlas TECHNICAL WHITE PAPER: COMPLETE LIST OF GENES CHARACTERIZED BY IN SITU HYBRIDIZATION IN ADULT HUMAN BRAIN STUDIES Table 1. Genes characterized by ISH in 1,000 gene survey in cortex (Cortex Study). Category Gene Symbol EntrezID Gene Description Gene Family Disease Comparative Marker Type Genomics A2M 2 alpha-2-macroglobulin extracellular Alzheimer’s VEC matrix AANAT 15 arylalkylamine N-acetyltransferase metabolic enzyme protein evolution AATF 26574 apoptosis antagonizing transcription transcription factor other factor neurodegenerative ABAT 18 4-aminobutyrate aminotransferase metabolic enzyme epilepsy interneuron ABCD1 215 ATP-binding cassette, sub-family D transporter other (ALD), member 1 neurodegenerative ACCN1 40 amiloride-sensitive cation channel 1, ion channel autism neuronal (degenerin) ACE 1636 angiotensin I converting enzyme metabolic enzyme Alzheimer’s (peptidyl-dipeptidase A) 1 ACHE 43 acetylcholinesterase (Yt blood group) metabolic enzyme Alzheimer’s OCTOBER 2012 v.2 alleninstitute.org Complete List of Genes Characterized by in situ Hybridization in Adult Human Brain Studies brain-map.org page 1 of 92 TECHNICAL WHITE PAPER ALLEN Human Brain Atlas Table 1. Genes characterized by ISH in 1,000 gene survey in cortex (Cortex Study). Category Gene Symbol EntrezID Gene Description Gene Family Disease Comparative Marker Type Genomics ACTB 60 actin, beta cytoskeletal mental retardation protein ACTN2 88 actinin, alpha 2 cytoskeletal schizophrenia protein ADAM23 8745 ADAM metallopeptidase domain 23 extracellular -
1 SUPPLEMENTARY TABLES Table S1. Membrane, Cytoskeletal, And
SUPPLEMENTARY TABLES Table S1. Membrane, cytoskeletal, and myofibrillar components that were co- purified with plakoglobin from skeletal muscle. # Unique Protein name Gene peptides Plakoglobin JUP 15 Sarcoglycan, delta SGCD 3 Laminin, beta 2 LAMB2 4 Laminin, gamma 1 LAMC1 17 Laminin Subunit 21 Beta 1 LAMB1 Spectrin alpha 2 SPNA2 7 Spectrin beta 1 SPNB1 8 Desmin DES 9 Plectin PLEC 36 Actinin, alpha 1 ACTN1 23 Actinin alpha 2 ACTN2 29 Actinin alpha 3 ACTN3 77 Actinin alpha 4 ACTN4 12 Plakophilin 1 PKP1 2 Desmoglein 1 DSG1C 2 Desmoplakin DSP 25 Cadherin 13 CDH13 2 Native protein assemblies of plakoglobin and bound proteins were isolated from membrane-cytoskeletal preparations of lower limb mouse muscles by size exclusion chromatography and anion-exchange column. Components that were co-purified with plakoglobin as a major protein peak were identified by mass spectrometry. 1 Table S2. Plakoglobin binds membrane, cytoskeletal, myofibrillar and insulin signaling components in skeletal muscle homogenates (6,000g supernatant). Gene # Unique Present in Protein name (NCBI) peptides high MW peak Plakoglobin JUP 16 + Dystrophin DMD 20 Sarcoglycan, alpha SGCA 3 Sarcoglycan, beta SGCB 2 Sarcoglycan, delta SGCD 2 + Alpha-1-syntrophin SNTA1 9 Beta-2-syntrophin SNTB2 4 Dystroglycan DAG1 3 Nitric oxide synthase 1 NOS1 3 Vinculin VCL 6 Caveolin 1 CAV1 5 Caveolin 3 CAV3 2 Laminin, beta 2 LAMB2 3 + Laminin, gamma 1 LAMC1 2 + Spectrin alpha 2 SPNA2 26 + Spectrin beta 1 SPNB1 13 + Insulin receptor substrate 1 IRS1 7 Insulin-like growth factor 2 receptor IGF2R 9 Insulin-like growth factor binding protein IGFALS 6 PI3K Catalytic Subunit Type 3 PIK3C3 1 PI3K Catalytic Subunit Gamma PIK3CG 2 PI3K-p85 PIK3R1 4 Desmin DES 26 + Plectin PLEC 147 + Actinin, alpha 1 ACTN1 6 + Actinin alpha 2 ACTN2 1 + Actinin alpha 3 ACTN3 2 + Actinin alpha 4 ACTN4 12 + Cadherin 13 CDH13 6 + Affinity purification of 6His-tagged plakoglobin and associated proteins from the soluble fraction of mouse lower limb muscles. -

Advancing Molecular Diagnostics in Sudden Unexplained Deaths
The author(s) shown below used Federal funding provided by the U.S. Department of Justice to prepare the following resource: Document Title: Advancing Molecular Diagnostics in Sudden Unexplained Deaths Author(s): Yingying Tang, M.D., Ph.D., DABMGG Document Number: 300769 Date Received: April 2021 Award Number: 2014-DN-BX-K001 This resource has not been published by the U.S. Department of Justice. This resource is being made publically available through the Office of Justice Programs’ National Criminal Justice Reference Service. Opinions or points of view expressed are those of the author(s) and do not necessarily reflect the official position or policies of the U.S. Department of Justice. Final Summary Overview NIJ FY 14 Basic Science Research to Support Forensic Science 2014-DN-BX-K001 Advancing Molecular Diagnostics in Sudden Unexplained Deaths March 28, 2021 Yingying Tang, MD, PhD, DABMGG Director, Molecular Genetics Laboratory Office of Chief Medical Examiner 421 East 26th Street New York, NY, 10016 Tel: 212-323-1340 Fax: 212-323-1540 Email: [email protected] This resource was prepared by the author(s) using Federal funds provided by the U.S. Department of Justice. Opinions or points of view expressed are those of the author(s) and do not necessarily reflect the official position or policies of the U.S. Department of Justice. Contents 1. PURPOSE OF THE PROJECT .............................................................................................................. 2 2. PROJECT DESIGN AND METHODS .................................................................................................... -
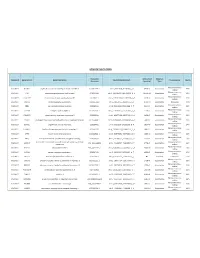
Somatic Mutations
SOMATIC MUTATIONS Transcript Amino Acid Mutation Sample ID Gene Symbol Gene Description Nucleotide (genomic) Consequence Mut % Accession (protein) Type Nonsynonymous PGDX11T ACSBG2 acyl-CoA synthetase bubblegum family member 2 CCDS12159.1 chr19_6141628_6141628_C_A 654A>D Substitution 46% coding Nonsynonymous PGDX11T ATR ataxia telangiectasia and Rad3 related CCDS3124.1 chr3_143721232_143721232_G_A 1451R>W Substitution 32% coding Nonsynonymous PGDX11T C1orf183 chromosome 1 open reading frame 183 CCDS841.1 chr1_112071336_112071336_C_T 224R>Q Substitution 22% coding PGDX11T CMYA5 cardiomyopathy associated 5 NM_153610 chr5_79063455_79063455_C_A 1037C>X Substitution Nonsense 34% Nonsynonymous PGDX11T CNR1 cannabinoid receptor 1 (brain) CCDS5015.1 chr6_88911646_88911646_C_T 23V>M Substitution 35% coding Nonsynonymous PGDX11T COL4A4 collagen; type IV; alpha 4 CCDS42828.1 chr2_227681807_227681807_G_A 227R>C Substitution 20% coding Nonsynonymous PGDX11T CYBASC3 cytochrome b; ascorbate dependent 3 CCDS8004.1 chr11_60877136_60877136_G_A 149R>C Substitution 34% coding Nonsynonymous PGDX11T DYRK3 dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 3 CCDS30999.1 chr1_204888091_204888091_G_A 309V>I Substitution 44% coding Nonsynonymous PGDX11T ELMO1 engulfment and cell motility 1 CCDS5449.1 chr7_37239295_37239295_G_A 160T>M Substitution 24% coding Nonsynonymous PGDX11T FAM83H family with sequence similarity 83; member H CCDS6410.2 chr8_144884473_144884473_C_A 90G>C Substitution 53% coding Nonsynonymous PGDX11T KPTN kaptin (actin binding protein)