Strand-Specific RNA-Seq Provides Greater Resolution of Transcriptome Profiling
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Proquesttm Pre-Made Cdna Libraries
Instruction Manual TM ProQuest Pre-made cDNA Libraries For detecting protein-protein interactions Version B December 12, 2002 25-0617 Table of Contents General Information ..............................................................2 Overview...............................................................................5 Using ProQuest™ Libraries....................................................8 pPC86.................................................................................13 pEXP-AD502.......................................................................15 Recipe.................................................................................17 Accessory Products ............................................................18 Purchaser Notification.........................................................19 Technical Service................................................................22 References..........................................................................24 1 General Information Contents 2 x 0.5 ml aliquots and Storage Each cDNA library is supplied in 80% SOB medium, 20% (v/v) glycerol. Store the library at -80°C. The cDNA library is stable for six months when stored properly. Titer of the Each library has greater than 5 x 109 cfu (colony Libraries forming units) derived from > 107 primary clones to ensure complete representation of rare sequences. ProQuest™ The following ProQuest™ Pre-made cDNA Libraries are Pre-made available from Invitrogen. For more information on cDNA preparing the library, see page -

In-Fusion Smarter Directional Cdna Library Construction Kit User Manual Table of Contents I
Takara Bio USA In-Fusion® SMARTer® Directional cDNA Library Construction Kit User Manual Cat. No. 634933 (050421) Takara Bio USA, Inc. 1290 Terra Bella Avenue, Mountain View, CA 94043, USA U.S. Technical Support: [email protected] United States/Canada Asia Pacific Europe Japan Page 1 of 40 800.662.2566 +1.650.919.7300 +33.(0)1.3904.6880 +81.(0)77.565.6999 In-Fusion SMARTer Directional cDNA Library Construction Kit User Manual Table of Contents I. List of Components .................................................................................................................................................. 4 II. Additional Materials Required .................................................................................................................................. 5 III. Introduction .......................................................................................................................................................... 6 IV. RNA Preparation & Handling ..............................................................................................................................11 A. Assessing the Quality of the RNA Template ........................................................................................................11 V. SMARTer cDNA Synthesis by LD-PCR .................................................................................................................13 A. Recommended Products ......................................................................................................................................13 -
Eukaryotic Non-Coding Rnas: New Targets for Diagnostics and Therapeutics?
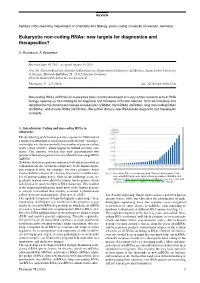
REVIEW Institute of Biochemistry, Department of Chemistry and Biology, Justus Liebig University of Giessen, Germany Eukaryotic non-coding RNAs: new targets for diagnostics and therapeutics? O. Rossbach, A. Bindereif Received June 30, 2015, accepted August 19, 2015 Prof. Dr. Albrecht Bindereif, Institute of Biochemistry, Department of Chemistry and Biology, Justus Liebig University of Giessen, Heinrich-Buff-Ring 58, 35392 Giessen, Germany [email protected] Pharmazie 71: 3–7 (2016) doi: 10.1691/ph.2016.5736 Non-coding RNAs (ncRNAs) in eukaryotes have recently developed to a very active research area in RNA biology, opening up new strategies for diagnosis and therapies of human disease. Here we introduce and describe the most important classes of eukaryotic ncRNAs: microRNAs (miRNAs), long non-coding RNAs (lncRNAs), and circular RNAs (circRNAs). We further discuss new RNA-based diagnostic and therapeutic concepts. 1. Introduction: Coding and non-coding RNAs in eukaryotes The deciphering of the human genome sequence in 2000 marked a unique breakthrough in modern molecular biology. An impor- tant insight was the unexpectedly low number of protein-coding genes (about 20,000), which lagged far behind previous esti- mates. This number revealed that only approximately two percent of the human genome is transcribed into messenger RNA (mRNA). However, the human genome sequence itself did not provide an explanation for the enormous complexity of the human organ- ism compared with, for example, the more primitive worm Caenorhabditis elegans (C. elegans) that carries a similar num- Fig. 1: Non-coding RNAs as an emerging field. The rapid development of the ber of protein-coding genes. -

A Pirna-Like Small RNA Interacts with and Modulates P-ERM Proteins in Human Somatic Cells
ARTICLE Received 18 Mar 2015 | Accepted 27 Apr 2015 | Published 22 June 2015 DOI: 10.1038/ncomms8316 OPEN A piRNA-like small RNA interacts with and modulates p-ERM proteins in human somatic cells Yuping Mei1, Yuyan Wang1,2, Priti Kumari3, Amol Carl Shetty3, David Clark1, Tyler Gable1, Alexander D. MacKerell4,5, Mark Z. Ma1, David J. Weber5,6, Austin J. Yang5, Martin J. Edelman5 & Li Mao1,5 PIWI-interacting RNAs (piRNAs) are thought to silence transposon and gene expression during development. However, the roles of piRNAs in somatic tissues are largely unknown. Here we report the identification of 555 piRNAs in human lung bronchial epithelial (HBE) and non-small cell lung cancer (NSCLC) cell lines, including 295 that do not exist in databases termed as piRNA-like sncRNAs or piRNA-Ls. Distinctive piRNA/piRNA-L expression patterns are observed between HBE and NSCLC cells. piRNA-like-163 (piR-L-163), the top down- regulated piRNA-L in NSCLC cells, binds directly to phosphorylated ERM proteins (p-ERM), which is dependent on the central part of UUNNUUUNNUU motif in piR-L-163 and the RRRKPDT element in ERM. The piR-L-163/p-ERM interaction is critical for p-ERM’s binding capability to filamentous actin (F-actin) and ERM-binding phosphoprotein 50 (EBP50). Thus, piRNA/piRNA-L may play a regulatory role through direct interaction with proteins in physiological and pathophysiological conditions. 1 Department of Oncology and Diagnostic Sciences, University of Maryland School of Dentistry, 650W Baltimore Street, Baltimore, Maryland 21201, USA. 2 Department of Thoracic Medical Oncology, Peking University Cancer Hospital and Institute, Beijing 100142, China. -

Library Construction and Screening
Library construction and screening • A gene library is a collection of different DNA sequences from an organism, • which has beenAlso called genomic libraries or gene banks. • cloned into a vector for ease of purification, storage and analysis. Uses of gene libraries • To obtain the sequences of genes for analysis, amplification, cloning, and expression. • Once the sequence is known probes, primers, etc. can be synthesized for further diagnostic work using, for example, hybridization reactions, blots and PCR. • Knowledge of a gene sequence also offers the possibility of gene therapy. • Also, gene expression can be used to synthesize a product in particular host cells, e.g. synthesis of human gene products in prokaryotic cells. two types of gene library depending upon the source of the DNA used. 1.genomic library. 2.cDNA library Types of GENE library: • genomic library contains DNA fragments representing the entire genome of an organism. • cDNA library contains only complementary DNA molecules synthesized from mRNA molecules in a cell. Genomic Library : • Made from nuclear DNA of an organism or species. • DNA is cut into clonable size pieces as randomly possible using restriction endonuclease • Genomic libraries contain whole genomic fragments including gene exons and introns, gene promoters, intragenic DNA,origins of replication, etc Construction of Genomic Libraries 1. Isolation of genomic DNA and vector. 2.Cleavage of Genomic DNA and vector by Restriction Endonucleases. 3.Ligation of fragmented DNA with the vector. 4.Transformation of -

Hammerhead Ribozymes Against Virus and Viroid Rnas
Hammerhead Ribozymes Against Virus and Viroid RNAs Alberto Carbonell, Ricardo Flores, and Selma Gago Contents 1 A Historical Overview: Hammerhead Ribozymes in Their Natural Context ................................................................... 412 2 Manipulating Cis-Acting Hammerheads to Act in Trans ................................. 414 3 A Critical Issue: Colocalization of Ribozyme and Substrate . .. .. ... .. .. .. .. .. ... .. .. .. .. 416 4 An Unanticipated Participant: Interactions Between Peripheral Loops of Natural Hammerheads Greatly Increase Their Self-Cleavage Activity ........................... 417 5 A New Generation of Trans-Acting Hammerheads Operating In Vitro and In Vivo at Physiological Concentrations of Magnesium . ...... 419 6 Trans-Cleavage In Vitro of Short RNA Substrates by Discontinuous and Extended Hammerheads ........................................... 420 7 Trans-Cleavage In Vitro of a Highly Structured RNA by Discontinuous and Extended Hammerheads ........................................... 421 8 Trans-Cleavage In Vivo of a Viroid RNA by an Extended PLMVd-Derived Hammerhead ........................................... 422 9 Concluding Remarks and Outlooks ........................................................ 424 References ....................................................................................... 425 Abstract The hammerhead ribozyme, a small catalytic motif that promotes self- cleavage of the RNAs in which it is found naturally embedded, can be manipulated to recognize and cleave specifically -

Global Tissue-Specific Transcriptome Analysis of Citrus Sinensis
www.nature.com/scientificdata opeN Global tissue-specifc transcriptome Data DeScrIpTor analysis of Citrus sinensis fruit across six developmental stages Received: 9 May 2019 Guizhi Feng, Juxun Wu & Hualin Yi Accepted: 23 July 2019 Published: xx xx xxxx Citrus sinensis fruit is a type of nonclimacteric fruit that mainly consists of four tissues: the epicarp, albedo, segment membrane and juice sac. The fruit quality is determined by the characteristics of these four tissues. However, our knowledge of the molecular processes that occur in these four tissues during citrus fruit development and ripening is limited. Tissue-specifc transcriptomes provide a comprehensive and detailed molecular regulatory network of citrus fruit development and ripening. In our study, we collected four types of tissue from C. sinensis fruits at six developmental stages. A total of 72 libraries were constructed from 24 samples (each sample had three replicates), and the transcriptomes were sequenced by an Illumina HiSeq 4000. The comprehensive analyses of the transcriptomes from the four tissues and six developmental stages presented here provide a valuable resource for the discovery of the molecular networks underlying citrus fruit development and ripening. Background & Summary Citrus, a nonclimacteric fruit, is widely cultivated worldwide. Citrus is mainly composed of the inedible epicarp (EP) and albedo (AL) and the edible segment membrane (SM) and juice sac (JS). Te development and ripening of citrus fruit is a complex and sophisticated regulatory process that can be divided into three stages: cell division stage; expansion stage involving cell enlargement and water, sugar accumulation; and ripening stage1. At present, most studies investigating citrus fruit development and ripening are based on the organ-wide or mixed-tissue level, which inevitably obscures many tissue-specifc phenomena. -

Questions with Answers- Nucleotides & Nucleic Acids A. the Components
Questions with Answers- Nucleotides & Nucleic Acids A. The components and structures of common nucleotides are compared. (Questions 1-5) 1._____ Which structural feature is shared by both uracil and thymine? a) Both contain two keto groups. b) Both contain one methyl group. c) Both contain a five-membered ring. d) Both contain three nitrogen atoms. 2._____ Which component is found in both adenosine and deoxycytidine? a) Both contain a pyranose. b) Both contain a 1,1’-N-glycosidic bond. c) Both contain a pyrimidine. d) Both contain a 3’-OH group. 3._____ Which property is shared by both GDP and AMP? a) Both contain the same charge at neutral pH. b) Both contain the same number of phosphate groups. c) Both contain the same purine. d) Both contain the same furanose. 4._____ Which characteristic is shared by purines and pyrimidines? a) Both contain two heterocyclic rings with aromatic character. b) Both can form multiple non-covalent hydrogen bonds. c) Both exist in planar configurations with a hemiacetal linkage. d) Both exist as neutral zwitterions under cellular conditions. 5._____ Which property is found in nucleosides and nucleotides? a) Both contain a nitrogenous base, a pentose, and at least one phosphate group. b) Both contain a covalent phosphodister bond that is broken in strong acid. c) Both contain an anomeric carbon atom that is part of a β-N-glycosidic bond. d) Both contain an aldose with hydroxyl groups that can tautomerize. ___________________________________________________________________________ B. The structures of nucleotides and their components are studied. (Questions 6-10) 6._____ Which characteristic is shared by both adenine and cytosine? a) Both contain one methyl group. -

Model-Based Integration of Genomics and Metabolomics Reveals SNP Functionality in Mycobacterium Tuberculosis
Model-based integration of genomics and metabolomics reveals SNP functionality in Mycobacterium tuberculosis Ove Øyåsa,b,1, Sonia Borrellc,d,1, Andrej Traunerc,d,1, Michael Zimmermanne, Julia Feldmannc,d, Thomas Liphardta,b, Sebastien Gagneuxc,d, Jörg Stellinga,b, Uwe Sauere, and Mattia Zampierie,2 aDepartment of Biosystems Science and Engineering, ETH Zurich, 4058 Basel, Switzerland; bSIB Swiss Institute of Bioinformatics, 1015 Lausanne, Switzerland; cDepartment of Medical Parasitoloy and Infection Biology, Swiss Tropical and Public Health Institute, 4051 Basel, Switzerland; dUniversity of Basel, 4058 Basel, Switzerland; and eInstitute of Molecular Systems Biology, ETH Zurich, 8093 Zurich, Switzerland Edited by Ralph R. Isberg, Tufts University School of Medicine, Boston, MA, and approved March 2, 2020 (received for review September 12, 2019) Human tuberculosis is caused by members of the Mycobacterium infection of macrophages (29–32). Beyond analyses of individual tuberculosis complex (MTBC) that vary in virulence and transmis- laboratory strains, however, no systematic characterization and sibility. While genome-wide association studies have uncovered comparative analysis of intrinsic metabolic differences across several mutations conferring drug resistance, much less is known human-adapted MTBC clinical strains has been performed. about the factors underlying other bacterial phenotypes. Variation If the metabolic and other phenotypic diversity between in the outcome of tuberculosis infection and diseases has been MTBC strains contributes to and modulates pathogenicity, an attributed primarily to patient and environmental factors, but obvious question is: Which elements of the limited genetic di- recent evidence indicates an additional role for the genetic diver- versity in the MTBC are responsible for phenotypic strain di- sity among MTBC clinical strains. -

IGA 8/E Chapter 8
8 RNA: Transcription and Processing WORKING WITH THE FIGURES 1. In Figure 8-3, why are the arrows for genes 1 and 2 pointing in opposite directions? Answer: The arrows for genes 1 and 2 indicate the direction of transcription, which is always 5 to 3. The two genes are transcribed from opposite DNA strands, which are antiparallel, so the genes must be transcribed in opposite directions to maintain the 5 to 3 direction of transcription. 2. In Figure 8-5, draw the “one gene” at much higher resolution with the following components: DNA, RNA polymerase(s), RNA(s). Answer: At the higher resolution, the feathery structures become RNA transcripts, with the longer transcripts occurring nearer the termination of the gene. The RNA in this drawing has been straightened out to illustrate the progressively longer transcripts. 3. In Figure 8-6, describe where the gene promoter is located. Chapter Eight 271 Answer: The promoter is located to the left (upstream) of the 3 end of the template strand. From this sequence it cannot be determined how far the promoter would be from the 5 end of the mRNA. 4. In Figure 8-9b, write a sequence that could form the hairpin loop structure. Answer: Any sequence that contains inverted complementary regions separated by a noncomplementary one would form a hairpin. One sequence would be: ACGCAAGCUUACCGAUUAUUGUAAGCUUGAAG The two bold-faced sequences would pair and form a hairpin. The intervening non-bold sequence would be the loop. 5. How do you know that the events in Figure 8-13 are occurring in the nucleus? Answer: The figure shows a double-stranded DNA molecule from which RNA is being transcribed. -

Mirna Detection Using a Rolling Circle Amplification and RNA
biosensors Article MiRNA Detection Using a Rolling Circle Amplification and RNA-Cutting Allosteric Deoxyribozyme Dual Signal Amplification Strategy Chenxin Fang, Ping Ouyang, Yuxing Yang, Yang Qing, Jialun Han, Wenyan Shang, Yubing Chen and Jie Du * State Key Laboratory of Marine Resource Utilization in South China Sea, College of Materials Science and Engineering, Hainan University, Haikou 570228, China; [email protected] (C.F.); [email protected] (P.O.); [email protected] (Y.Y.); [email protected] (Y.Q.); [email protected] (J.H.); [email protected] (W.S.); [email protected] (Y.C.) * Correspondence: [email protected] Abstract: A microRNA (miRNA) detection platform composed of a rolling circle amplification (RCA) system and an allosteric deoxyribozyme system is proposed, which can detect miRNA-21 rapidly and efficiently. Padlock probe hybridization with the target miRNA is achieved through complementary base pairing and the padlock probe forms a closed circular template under the action of ligase; this circular template results in RCA. In the presence of DNA polymerase, RCA proceeds and a long chain with numerous repeating units is formed. In the presence of single-stranded DNA (H1 and H2), multi-component nucleic acid enzymes (MNAzymes) are formed that have the ability to cleave substrates. Finally, substrates containing fluorescent and quenching groups and magnesium ions are added to the system to activate the MNAzyme and the substrate cleavage reaction, thus achieving fluorescence intensity amplification. The RCA–MNAzyme system has dual signal amplification and presents a sensing platform that demonstrates broad prospects in the analysis and detection of Citation: Fang, C.; Ouyang, P.; Yang, nucleic acids. -

Comparative Analyses of Long Non-Coding RNA in Lean and Obese Pigs
www.impactjournals.com/oncotarget/ Oncotarget, 2017, Vol. 8, (No. 25), pp: 41440-41450 Research Paper Comparative analyses of long non-coding RNA in lean and obese pigs Lin Yu1, Lina Tai1, Lifang Zhang1, Yi Chu1, Yixing Li1 and Lei Zhou1 1State Key Laboratory for Conservation and Utilization of Subtropical Agro-Bioresources, College of Animal Science and Technology, Guangxi University, Nanning, P.R. China Correspondence to: Yixing Li, email: [email protected] Lei Zhou, email: [email protected] Keywords: lncRNA, pig, obesity, QTL Received: April 28, 2017 Accepted: May 15, 2017 Published: May 26, 2017 Copyright: Yu et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License 3.0 (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Objectives: Current studies have revealed that long non-coding RNA plays a crucial role in fat metabolism. However, the difference of lncRNA between lean (Duroc) and obese (Luchuan) pig remain undefined. Here, we investigated the expressional profile of lncRNA in these two pigs and discussed the relationship between lncRNA and fat deposition. Materials and Methods: The Chinese Luchuan pig has a dramatic differences in backfat thickness as compared with Duroc pig. In this study, 4868 lncRNA transcripts (including 3235 novel transcripts) were identified. We determined that patterns of differently expressed lncRNAs and mRNAs are strongly tissue-specific. The differentially expressed lncRNAs in adipose tissue have 794 potential target genes, which are involved in adipocytokine signaling pathways, the PI3k-Akt signaling pathway, and calcium signaling pathways.