An Integrated Semiconductor Device Enabling Non-Optical Genome Sequencing
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ion-Sensitive Field Effect Transistor (ISFET) for MEMS Multisensory Chips at RIT Murat Baylav
Rochester Institute of Technology RIT Scholar Works Theses Thesis/Dissertation Collections 2010 Ion-sensitive field effect transistor (ISFET) for MEMS multisensory chips at RIT Murat Baylav Follow this and additional works at: http://scholarworks.rit.edu/theses Recommended Citation Baylav, Murat, "Ion-sensitive field effect transistor (ISFET) for MEMS multisensory chips at RIT" (2010). Thesis. Rochester Institute of Technology. Accessed from This Thesis is brought to you for free and open access by the Thesis/Dissertation Collections at RIT Scholar Works. It has been accepted for inclusion in Theses by an authorized administrator of RIT Scholar Works. For more information, please contact [email protected]. Title Page Ion-Sensitive Field Effect Transistor (ISFET) for MEMS Multisensory Chips at RIT By Murat Baylav A Thesis Submitted In Partial Fulfillment of the Requirements of the Degree of Master of Science in Microelectronic Engineering Approved by: Professor ___________________________________ Date: __________________ Dr. Lynn F. Fuller (Thesis Advisor) Professor ___________________________________ Date: __________________ Dr. Karl D. Hirschman (Thesis Committee Member) Professor ___________________________________ Date: __________________ Dr. Santosh K. Kurinec (Thesis Committee Member) Professor ___________________________________ Date: __________________ Dr. Robert Pearson (Director, Microelectronic Engineering Program) Professor ___________________________________ Date: __________________ Dr. Sohail Dianat (Chair, Electrical -

Temperature Compensation Circuit for ISFET Sensor
Journal of Low Power Electronics and Applications Article Temperature Compensation Circuit for ISFET Sensor Ahmed Gaddour 1,2,* , Wael Dghais 2,3, Belgacem Hamdi 2,3 and Mounir Ben Ali 3,4 1 National Engineering School of Monastir (ENIM), University of Monastir, Monastir 5000, Tunisia 2 Electronics and Microelectronics Laboratory, LR99ES30, Faculty of Sciences of Monastir, University of Monastir, Monastir 5000, Tunisia; [email protected] (W.D.); [email protected] (B.H.) 3 Higher Institute of Applied Sciences and Technology of Sousse (ISSATSo), University of Sousse, Sousse 4003, Tunisia; [email protected] 4 Nanomaterials, Microsystems for Health, Environment and Energy Laboratory, LR16CRMN01, Centre for Research on Microelectronics and Nanotechnology, Sousse 4034, Tunisia * Correspondence: [email protected]; Tel.: +216-50998008 Received: 3 November 2019; Accepted: 21 December 2019; Published: 4 January 2020 Abstract: PH measurements are widely used in agriculture, biomedical engineering, the food industry, environmental studies, etc. Several healthcare and biomedical research studies have reported that all aqueous samples have their pH tested at some point in their lifecycle for evaluation of the diagnosis of diseases or susceptibility, wound healing, cellular internalization, etc. The ion-sensitive field effect transistor (ISFET) is capable of pH measurements. Such use of the ISFET has become popular, as it allows sensing, preprocessing, and computational circuitry to be encapsulated on a single chip, enabling miniaturization and portability. However, the extracted data from the sensor have been affected by the variation of the temperature. This paper presents a new integrated circuit that can enhance the immunity of ion-sensitive field effect transistors (ISFET) against the temperature. -

An Extended CMOS ISFET Model Incorporating the Physical Design
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Spiral - Imperial College Digital Repository IEEE TRANSACTIONS ON ELECTRON DEVICES, VOL. XXX, NO. XXX, XXX XXX 1 An Extended CMOS ISFET Model Incorporating the Physical Design Geometry and the Effects on Performance and Offset Variation Yan Liu, Student Member, IEEE, Pantelis Georgiou, Member, IEEE, Themistoklis Prodromakis, Member, IEEE, Timothy G. Constandinou, Senior Member, and Christofer Toumazou, Fellow, IEEE Abstract—This paper presents an extended model for the as large-scale, highly-integrated chemical sensor arrays [4], CMOS-based Ion-Sensitive-Field-Effect-Transistor (ISFET), in- [6]. corporating design parameters associated with the physical In this paper we present an extended model for CMOS- geometry of the device. This can, for the first time, provide a good match between calculated and measured characteristics by based ISFETs to include both the first order effects, (i.e. taking into account the effects of non-idealities such as threshold intrinsic dimension-related characteristics) and second order voltage variation and sensor noise. The model is evaluated effects, (i.e. non-linear characteristics). By focusing on the through a number of devices with varying design parameters effect of varying the design parameters (i.e physical dimen- (chemical sensing area and MOSFET dimensions) fabricated in sions) a capacitance-based model is derived which includes all a commercially-available 0.35µm CMOS technology. Threshold voltage, subthreshold slope, chemical sensitivity, drift and noise capacitive structures, the values of which are directly related were measured and compared to the simulated results. The to physical dimensions. -

Dynamic Ph Sensor with Embedded Calibration Scheme by Advanced CMOS Finfet Technology
sensors Article Dynamic pH Sensor with Embedded Calibration Scheme by Advanced CMOS FinFET Technology Chien-Ping Wang 1, Ying-Chun Shen 2, Peng-Chun Liou 1, Yu-Lun Chueh 2, Yue-Der Chih 3, Jonathan Chang 3, Chrong-Jung Lin 1 and Ya-Chin King 1,* 1 Institute of Electronics Engineering, National Tsing Hua University, Hsinchu 30013, Taiwan; [email protected] (C.-P.W.); [email protected] (P.-C.L.); [email protected] (C.-J.L.) 2 Institute of Materials Science and Engineering, National Tsing Hua University, Hsinchu 30013, Taiwan; [email protected] (Y.-C.S.); [email protected] (Y.-L.C.) 3 Design Technology Division, Taiwan Semiconductor Manufacturing Company, Hsinchu 30075, Taiwan; [email protected] (Y.-D.C.); [email protected] (J.C.) * Correspondence: [email protected]; Tel.: +886-3-5162219 or +886-3-5721804 or +1-123914307 Received: 4 March 2019; Accepted: 29 March 2019; Published: 2 April 2019 Abstract: In this work, we present a novel pH sensor using efficient laterally coupled structure enabled by Complementary Metal-Oxide Semiconductor (CMOS) Fin Field-Effect Transistor (FinFET) processes. This new sensor features adjustable sensitivity, wide sensing range, multi-pad sensing capability and compatibility to advanced CMOS technologies. With a self-balanced readout scheme and proposed corresponding circuit, the proposed sensor is found to be easily embedded into integrated circuits (ICs) and expanded into sensors array. To ensure the robustness of this new device, the transient response and noise analysis are performed. In addition, an embedded calibration operation scheme is implemented to prevent the proposed sensing device from the background offset from process variation, providing reliable and stable sensing results. -

Vlsi Design Lecture Notes B.Tech (Iv Year – I Sem) (2018-19)
VLSI DESIGN LECTURE NOTES B.TECH (IV YEAR – I SEM) (2018-19) Prepared by Dr. V.M. Senthilkumar, Professor/ECE & Ms.M.Anusha, AP/ECE Department of Electronics and Communication Engineering MALLA REDDY COLLEGE OF ENGINEERING & TECHNOLOGY (Autonomous Institution – UGC, Govt. of India) Recognized under 2(f) and 12 (B) of UGC ACT 1956 (Affiliated to JNTUH, Hyderabad, Approved by AICTE - Accredited by NBA & NAAC – ‘A’ Grade - ISO 9001:2015 Certified) Maisammaguda, Dhulapally (Post Via. Kompally), Secunderabad – 500100, Telangana State, India Unit -1 IC Technologies, MOS & Bi CMOS Circuits Unit -1 IC Technologies, MOS & Bi CMOS Circuits UNIT-I IC Technologies Introduction Basic Electrical Properties of MOS and BiCMOS Circuits MOS I - V relationships DS DS PMOS MOS transistor Threshold Voltage - VT figure of NMOS merit-ω0 Transconductance-g , g ; CMOS m ds Pass transistor & NMOS Inverter, Various BiCMOS pull ups, CMOS Inverter Technologies analysis and design Bi-CMOS Inverters Unit -1 IC Technologies, MOS & Bi CMOS Circuits INTRODUCTION TO IC TECHNOLOGY The development of electronics endless with invention of vaccum tubes and associated electronic circuits. This activity termed as vaccum tube electronics, afterward the evolution of solid state devices and consequent development of integrated circuits are responsible for the present status of communication, computing and instrumentation. • The first vaccum tube diode was invented by john ambrase Fleming in 1904. • The vaccum triode was invented by lee de forest in 1906. Early developments of the Integrated Circuit (IC) go back to 1949. German engineer Werner Jacobi filed a patent for an IC like semiconductor amplifying device showing five transistors on a common substrate in a 2-stage amplifier arrangement. -

A Novel Coding Method for Radar Signal Waveform Design
APPLICATION OF ION SELECTIVE FIELD EFFECT TRANSISTOR FOR HYDROGEN ION MEASUREMENT SYSTEM Ibrahim Elkhier Hussien Arfeen1, Abdul Rasoul Jabar Kizar Alzubaidi2 1 Sudanese Standards and Metrology Organization, Port Sudan 2 Electronic Dept- Engineering College-Sudan University for science and Technology Received December 2010, accepted after revision June 2011 ُمـ ْســتَ ْخـلـَص َهذف هذا انبحث إنً دراصت تزانزصتىر تأثُز انًدبل كًحضبس كًُُبئٍ َضتخذو نًزاقبت اﻷس انهُذروخُنٍ فٍ يدبل انكًُُبء انتحهُهُت و انبُئُت. تعتًذ حضبصُت حضبس تزانزصتىر تأثُز انًدبل عهٍ نىع انًبدة اﻹنكتزونُت انًضتخذيت كًدش فٍ بىابت انتزانزصتىر. وَضتخذو فٍ يىاضع انقُبس انتٍ َصعب فُهب اصتخذاو انًحضبس انزخبخٍ انقببم نهكضز. تضتخذو دائزة إنكتزونُت نزبط يحضبس تزانزصتىر تأثُز انًدبل تًكنه ين انعًم بصىرة فعبنت فٍ انًذي ين اﻷس انهُذروخُنٍ 4 إنً 44. وظُفت انذائزة اﻹنكتزونُت هي تحىَم تُبر دخم انًحضبس انذٌ ًَثم قُى اﻷس انهُذروخنٍ إنً خهذ خزج يتنبصب. تى اختببر انتصًُى بىاصطت يحبنُم قُبصُت وخًضت أنىاع ين انًحبنُم انًبئُت وتى انحصىل عهً اننتبئح انًطهىبت. تى رصى انعﻻقت بُن اننتبئح انًتحصم عهُهب بىاصطت انذائزة اﻹنكتزونُت وقُى اﻷس انهُذروخُنٍ نبعض انًحبنُم انًبئُت. ABSTRACT This paper aims to study field effect transistors as chemical sensor used for monitoring hydrogen ion concentration (H+) measurement in chemical analysis and environment fields, to detect the pH of the subjected solution. The Ion-Selective Field Effect Transistor (ISFET) sensor sensitivity depends mainly on the choice of the gate dielectric material (Silicon Nitride Membrane). The Ion-Selective Field Effect Transistor is (ISFET) used in places where fragile glass electrodes will be damaged. The interface circuit supplies the sensor with a constant current and voltage. The pH value is identified from the output voltage of the circuit. -

Design of Low Noise Ph-ISFET Microsensors and Integrated Suspended Inductors with Increased Quality B
Design of low noise pH-ISFET microsensors and integrated suspended inductors with increased quality B. Palan To cite this version: B. Palan. Design of low noise pH-ISFET microsensors and integrated suspended inductors with increased quality. Micro and nanotechnologies/Microelectronics. Institut National Polytechnique de Grenoble - INPG, 2002. English. tel-00003085 HAL Id: tel-00003085 https://tel.archives-ouvertes.fr/tel-00003085 Submitted on 4 Jul 2003 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. ÁÆËÌÁÌÍÌ ÆÌÁÇÆÄ ÈÇÄ Ì ÀÆÁÉÍ Ê ÆÇÄ Ó Æ ØØÖÙ ÔÖ Ð ÐÓØ ÕÙ ÌÀ Ë Æ ÇÌÍÌ ÄÄ ÔÓÙÖ ÓØÒÖ Ð Ö ÇÌÍÊ Ð³ÁÆÈ Ì Ä³ÍÆÁÎÊËÁÌ ÌÀÆÁÉÍ ÌÀÉÍ ÈÊ Í ËÔÐØ ÅÖÓÐØÖÓÒÕÙ ÔÖÔÖ Ù Ð ÓÖØÓÖ ÌÁÅ Ò× Ð Ö Ð³!ÓÐ Ó !ØÓÖÐ ÄÌÊÇÆÁÉ͸ ÄÌÊÇÌÀÆÁÉ͸ ÍÌÇÅ ÌÁÉ͸ ÌÄÇÅÅÍÆÁ ÌÁÇÆ˸ ËÁÆ Ä Ø Ù ÔÖØÑÒØ Ð ÅÖÓÐØÖÓÒÕÙ¸ ÍÒÚÖ×Ø ÌÒÕÙ ÌÕÙ ´ÍÌ̵ ÈÖÙ ÔÖ×ÒØ Ø ×ÓÙØÒÙ ÔÙÐÕÙÑÒØ ÔÖ ÓÙ×ÐÚ È Ä Æ Ð ½ÑÖ× ¾¼¼¾ ÌØÖ ÇÆÈÌÁÇÆ ÅÁÊÇ ÈÌÍÊË ÔÀ¹ÁËÌ ÁÄ ÊÍÁÌ Ì ³ÁÆÍÌ ÆË ÁÆÌÊË ËÍËÈÆÍË ÇÊÌ ÌÍÊ ÉÍ ÄÁÌ É ß Ö !Ø ÙÖ× % Ø × & Ó ØÙÖ ÖÒÖ ÇÍÊÌÇÁË ÈÖÓ××ÙÖ ÅÖÓ×ÐÚÀÍËÃ ß ÂÍÊ( ź ÈÖÖ ÆÌÁÄ ¸ÈÖ×ÒØ Åº ÖÒÖ ÇÍÊÌÇÁË ¸ÖØÙÖ Ø× ÌÁÅ -
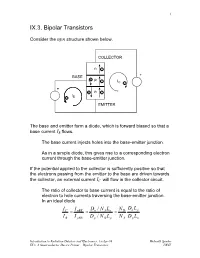
IX.3. a Semiconductor Device Primer – Bipolar Transistors LBNL 2
1 IX.3. Bipolar Transistors Consider the npn structure shown below. COLLECTOR n- BASE + +p -IC- + +n- I -B EMITTER The base and emitter form a diode, which is forward biased so that a base current IB flows. The base current injects holes into the base-emitter junction. As in a simple diode, this gives rise to a corresponding electron current through the base-emitter junction. If the potential applied to the collector is sufficiently positive so that the electrons passing from the emitter to the base are driven towards the collector, an external current IC will flow in the collector circuit. The ratio of collector to base current is equal to the ratio of electron to hole currents traversing the base-emitter junction. In an ideal diode IC I nBE Dn / N ALn N D Dn Lp = = = I B I pBE Dp / N D Lp N A Dp Ln Introduction to Radiation Detctors and Electronics, 13-Apr-99 Helmuth Spieler IX.3. A Semiconductor Device Primer – Bipolar Transistors LBNL 2 If the ratio of doping concentrations in the emitter and base regions ND /NA is sufficiently large, the collector current will be greater than the base current. ⇒ DC current gain Furthermore, we expect the collector current to saturate when the collector voltage becomes large enough to capture all of the minority carrier electrons injected into the base. Since the current inside the transistor comprises both electrons and holes, the device is called a bipolar transistor. Dimensions and doping levels of a modern high-frequency transistor (5 – 10 GHz bandwidth) 0 0.5 1.0 1.5 Distance [µm] (adapted from Sze) Introduction to Radiation Detctors and Electronics, 13-Apr-99 Helmuth Spieler IX.3. -

Laboratory Exercise 2 DC Characteristics of Bipolar Junction
DEPARTMENT OF SEMICONDUCTOR AND OPTOELECTRONIC DEVICES Semiconductor Device Laboratory Laboratory Exercise 2 DC characteristics of Bipolar Junction Transistors (BJT) The aim of the exercise The main aim of this laboratory exercise is to understand principles of operation of Bipolar Junction Transistors (BJT). It covers the measurements of static and small signal parameters. Backgrounds Physical structure of the BJT BJT is a semiconductor device having a three-layer structure with three external electrodes, the emitter (E), the base (B), and the collector (C). As shown in Fig. 1, the structure may be p-n-p or n-p-n type. Despite of the transistor type the emitter layer has always more acceptor or donor impurities added than the base or the collector layer. This asymmetry results from different roles the emitter and the collector layers play in the BJT. In normal operation of the BJT (as an amplifier), the base-emitter junction is forward biased and the base-collector junction is reverse biased. Transistor amplification is controlled by changing the current flow through the base-emitter junction. C N C . C P C . P B. N B. B . B . E . N . E P . E E (a) (b) Fig. 1. The n-p-n BJT (a) and the p-n-p BJT (b) along with their symbols There are three operating configurations of the BJT. These are the common-emitter (OE) configuration, the common-base configuration (OB), and the common-collector (OC) configuration. These configurations are shown in Fig. 2. (a) (b) (c) Fig. 2 The n-p-n transistor operating configurations: (a) common-emitter, (b) common-base, (c) common-collector DC characteristics Four types of characteristics can be defined for each of the transistor operating configurations. -

Field Effect Transistor (FET) Types and Features
Field Effect Transistor (FET) Types and Features Here I’m discussing about the topic FET. FET is another semiconductor device like BJT which can be used as switch, amplifier, resistor etc. FET consists of 3 terminals. Drain(D) Source(S) Gate(G) In these 3 terminals, Gate terminal acts as a controlling terminal. We know that a BJT acts as a current controlling device. Like that, FET also acts as a voltage controlling device. Here, the voltage between gate and source controls the drain current. So, it is called as voltage controlled device. FET Features: FET is more temperature stable compared to BJT It requires less space compared to BJT. So it is used heavily in circuits. FET has higher input impedance. So, it is more useful in amplifiers. Types of FETs: 1. Junction FET 2. MESFET 3. MOSFET Here I am discussing about the topic JFET. Junction FET (JFET): Basically the JFET is classified into 2 types. N-Channel JFET P-Channel JFET N-Channel JFET: When we consider a silicon bar and fabricated n-type at its two ends and heavily doped p-type materials at each side of the bar, the thin region will be remained as observed in figure is channel. Since this channel is in n-type bar this is called as n-channel FET. Here the current is carried by electrons. P-Channel FET: When we consider a silicon bar and fabricated p-type at its two ends and heavily doped n-type materials at each side of the bar, here the thin region remained as observed in figure is channel. -

Csd25301w1015
P-Channel CICLON NexFET™ Power MOSFETs CSD25301W1015 Features Product Summary D D • Ultra Low Qg & Qgd VDS -20 V Q 2.0 nC • Small Footprint g S S Qgd 0.32 nC • Low Profile 0.65mm height VGS= -1.5V 175 mΩ • Pb Free S G RDS(on) VGS=-2.5V 80 mΩ VGS=-4.5V 62 mΩ RoHS Compliant • CSP 1.0 x 1.5 mm Wafer Vth -0.75 V • Halogen Free Level Package Top View o Maximum Values (TA=25 C unless otherwise stated) Symbol Parameter Value Units VDS Drain to Source Voltage -20 V VGS Gate to Source Voltage ±8 V 1 ID Continuous Drain Current, TJ = 25°C -2.2 A IDM Pulsed Drain Current, TJ = 25°C1,2 -8.8 A 1 PD Power Dissipation 1.5 W TJ, TSTG Operating Junction and Storage Temperature Range -55 to 150 °C 0 1. RthJA = 85 C/W on max Cu (2 oz.) on 0.060” thick FR4 PCB 2. Pulse width ≤300 µs, duty cycle ≤ 2% RDS(ON) vs. VGS Gate Charge 300 6 ID = -1A V DS = -10V ) 250 5 ID = -1A Ω T = 125ºC 200 J 4 TJ = 25ºC 150 3 100 (V) Voltage - Gate 2 - On Resistance (m GS -V DS(on) 50 R 1 0 0 0123456 0 0.25 0.5 0.75 1 1.25 1.5 1.75 2 2.25 2.5 -V - Gate to Source Voltage (V) Qg - Gate Charge (nC) GS Ordering Information Type Package Package Media Qty Ship CSD25301W1015 1.0 X 1.5 Wafer Level Package 7 inch reel 3000 Tape and Reel © 2008 CICLON Semiconductor Device Corp., rev 2.3 www.ciclonsemi.com All rights reserved. -

An Ion-Gated Bipolar Amplifier for Ion Sensing with Enhanced Signal and Improved Noise Performance
An ion-gated bipolar amplifier for ion sensing with enhanced signal and improved noise performance Da Zhang, Xindong Gao, Si Chen, Hans Norström, Ulf Smith, Paul Solomon, Shi-Li Zhang, and Zhen Zhang Citation: Applied Physics Letters 105, 082102 (2014); doi: 10.1063/1.4894240 View online: http://dx.doi.org/10.1063/1.4894240 View Table of Contents: http://scitation.aip.org/content/aip/journal/apl/105/8?ver=pdfcov Published by the AIP Publishing Articles you may be interested in Large area graphene ion sensitive field effect transistors with tantalum pentoxide sensing layers for pH measurement at the Nernstian limit Appl. Phys. Lett. 105, 083101 (2014); 10.1063/1.4894078 Hydrogen ion-selective electrolyte-gated organic field-effect transistor for pH sensing Appl. Phys. Lett. 104, 193305 (2014); 10.1063/1.4878539 Nanodiamond-gated silicon ion-sensitive field effect transistor Appl. Phys. Lett. 98, 153507 (2011); 10.1063/1.3568887 Nanodiamond-gated diamond field-effect transistor for chemical sensing using hydrogen-induced transfer doping for channel formation Appl. Phys. Lett. 97, 203503 (2010); 10.1063/1.3518060 Theoretical study of electrolyte gate Al Ga N Ga N field effect transistors J. Appl. Phys. 97, 033703 (2005); 10.1063/1.1847730 This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP: 130.238.20.83 On: Wed, 27 Aug 2014 12:08:59 APPLIED PHYSICS LETTERS 105, 082102 (2014) An ion-gated bipolar amplifier for ion sensing with enhanced signal and improved noise performance Da Zhang, Xindong Gao, Si Chen,a) Hans Norstrom,€ Ulf Smith, Paul Solomon,b) Shi-Li Zhang, and Zhen Zhangc) Solid-State Electronics, The A˚ngstrom€ Laboratory, Uppsala University, SE-751 21 Uppsala, Sweden (Received 30 July 2014; accepted 16 August 2014; published online 25 August 2014) This work presents a proof-of-concept ion-sensitive device operating in electrolytes.