Thesis Title
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

United States Patent 19 11 Patent Number: 5,418,243 Angerbauer Et Al
USOO5418243A United States Patent 19 11 Patent Number: 5,418,243 Angerbauer et al. 45 Date of Patent: May 23, 1995 54 SUBSTITUTED 4-PHENYL-PYRIDONES 4,215,126 7/1980. Durant et al........................ 514/345 AND 4-PHENYL-3-ALKOXYPYRIDNES 4,684,477 8/1987 Sugimori et al. ... 546/290 4,916,239 4/1990 Treiber ................. ... 549/292 75 Inventors: Rolf Angerbauer; Peter Fey; Walter 4,988,711 1/1991 Angerbauer et al. ... 514/326 Hibsch, all of Wuppertal; Thomas 5,032,602 7/1991 Fey et al. ................. ... 54/345 Philipps, Cologne; Hilmar Bischoff, 5,064,841 11/1991 Angerbauer et al. ... 514/336 Wuppertal; Hans-Peter Krause, 5,138,090 8/1992 Fey et al. .............................. 560/59 Schwelm; Jörg Peterson-von Gehr, Bochum; Delf Schmidt, Wuppertal, FOREIGN PATENT DOCUMENTS all of Germany 0373423 6/1990 European Pat. Off. 73 Assignee: Bayer Aktiengesellschaft, OTHER PUBLICATIONS Leverkusen, Germany J. Med. Chem., 1990, vol. 33, pp. 52-60; “Synthesis and 21) Appl. No.: 166,775 Biological Activity of New HGM-CoA Reductase 22 Filed: Dec. 14, 1993 Inhibitors ... ', G. Beck. Primary Examiner-C. Warren Ivy (30) Foreign Application Priority Data Assistant Examiner-A. A. Owens Dec. 21, 1992 DE Germany ........................ 4243 278.2 Attorney, Agent, or Firm-Sprung, Horn, Kramer & Jun. 28, 1993 DE Germany ........................ 43 21421.5 Woods 511 Int. Cl...................... A61K 31/44; C07D 213/64 57 ABSTRACT 52 U.S. C. ...................................... 514/345; 546/24; 546/290; 514/89 Substituted 4-phenyl-pyridones and 4-phenyl-2-alkox 58 Field of Search .................. 546/290, 24; 514/345, ypyridines are prepared by reducing corresponding 514/89 4-phenyl-pyridone and 4-phenyl-2-alkoxypyridine de rivatives. -

Beryllium Chloride Apparatus. Figure M
Durham E-Theses Organic and hydride chemistry of beryllium Bell, N.A How to cite: Bell, N.A (1964) Organic and hydride chemistry of beryllium, Durham theses, Durham University. Available at Durham E-Theses Online: http://etheses.dur.ac.uk/8894/ Use policy The full-text may be used and/or reproduced, and given to third parties in any format or medium, without prior permission or charge, for personal research or study, educational, or not-for-prot purposes provided that: • a full bibliographic reference is made to the original source • a link is made to the metadata record in Durham E-Theses • the full-text is not changed in any way The full-text must not be sold in any format or medium without the formal permission of the copyright holders. Please consult the full Durham E-Theses policy for further details. Academic Support Oce, Durham University, University Oce, Old Elvet, Durham DH1 3HP e-mail: [email protected] Tel: +44 0191 334 6107 http://etheses.dur.ac.uk ORGANIC AND HYDRIDE CHEMISTRY OF BERYLLIUM oy N.A. BELL. A thesis.submitted for the Degree of Doctor of Philosophy in the University- of Durham. June 196^-. I Acknowledgements. The author wishes to express his sincere thanks to Professor G-.E. Coates, M.A., D.Sc., F.R.I.C., under whose direction this research was carried out, for his constant encouragement and extremely valuable advice. ' The author is also indebted to the Department of Scien• tific and Industrial Research for a Research StudiSntshrp. I I Memorandum. The work described in this thesis was carried out in the University of Durham between September 19b1 and May 196A-. -

Sodium Aluminum Hydride
Sodium aluminum hydride sc-251007 Material Safety Data Sheet Hazard Alert Code Key: EXTREME HIGH MODERATE LOW Section 1 - CHEMICAL PRODUCT AND COMPANY IDENTIFICATION PRODUCT NAME Sodium aluminum hydride STATEMENT OF HAZARDOUS NATURE CONSIDERED A HAZARDOUS SUBSTANCE ACCORDING TO OSHA 29 CFR 1910.1200. NFPA FLAMMABILITY3 HEALTH3 HAZARD INSTABILITY1 W SUPPLIER Santa Cruz Biotechnology, Inc. 2145 Delaware Avenue Santa Cruz, California 95060 800.457.3801 or 831.457.3800 EMERGENCY: ChemWatch Within the US & Canada: 877-715-9305 Outside the US & Canada: +800 2436 2255 (1-800-CHEMCALL) or call +613 9573 3112 SYNONYMS Al-H4.Na, "aluminate (1-), tetrahydro-, sodium", "aluminum sodium hydride", "sodium aluminium tetrahydride", "sodium tetrahydroaluminate (1-)" Section 2 - HAZARDS IDENTIFICATION CHEMWATCH HAZARD RATINGS Min Max Flammability: 3 Toxicity: 2 Body Contact: 3 Min/Nil=0 Low=1 Reactivity: 1 Moderate=2 High=3 Chronic: 2 Extreme=4 CANADIAN WHMIS SYMBOLS 1 of 7 EMERGENCY OVERVIEW RISK Contact with water liberates extremely flammable gases. Harmful if swallowed. Causes burns. Risk of serious damage to eyes. Highly flammable. POTENTIAL HEALTH EFFECTS ACUTE HEALTH EFFECTS SWALLOWED ! The material can produce chemical burns within the oral cavity and gastrointestinal tract following ingestion. ! Accidental ingestion of the material may be harmful; animal experiments indicate that ingestion of less than 150 gram may be fatal or may produce serious damage to the health of the individual. EYE ! The material can produce chemical burns to the eye following direct contact. Vapors or mists may be extremely irritating. ! If applied to the eyes, this material causes severe eye damage. SKIN ! The material can produce chemical burns following direct contactwith the skin. -
DG Classification.Pdf
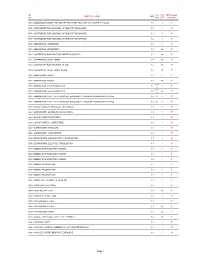
UN Sub PSA MPA weight CHEMICAL NAME IMO No Risk GRP restriction 0004 AMMONIUM PICRATE, DRY OR WETTED WITH LESS THAN 10% WATER, BY MASS 1.1 1 Y* 0005 CARTRIDGES FOR WEAPONS, WITH BURSTING CHARGE 1.1 1 Y* 0006 CARTRIDGES FOR WEAPONS, WITH BURSTING CHARGE 1.1 1 Y* 0007 CARTRIDGES FOR WEAPONS, WITH BURSTING CHARGE 1.2 1 Y* 0009 AMMUNITION, INCENDIARY, 1.2 1 Y* 0010 AMMUNITION, INCENDIARY, 1.3 2A Y 0012 CARTRIDGES FOR WEAPONS, INERT PROJECTILE 1.4 2A Y 0012 CARTRIDGES, SMALL ARMS 1.4 2A Y 0014 CARTRIDGES FOR WEAPONS, BLANK 1.4 2A Y 0014 CARTRIDGES, SMALL ARMS, BLANK 1.4 2A Y 0015 AMMUNITION, SMOKE, 1.2 1 Y* 0016 AMMUNITION, SMOKE, 1.3 2A Y 6.1 / 0018 AMMUNITION, TEAR-PRODUCING, 1.2 1 Y* 8 6.1 / 0019 AMMUNITION, TEAR-PRODUCING, 1.3 2A Y 8 0020 AMMUNITION, TOXIC, WITH BURSTER, EXPELLING CHARGE OR PROPELLING CHARGE 1.2 6.1 1 Y* 0021 AMMUNITION, TOXIC, WITH BURSTER, EXPELLING CHARGE OR PROPELLING CHARGE 1.3 6.1 1 Y* 0027 BLACK POWDER, GRANULAR, OR AS MEAL 1.1 1 Y* 0027 GUNPOWDER, GRANULAR, OR AS A MEAL 1.1 1 Y* 0028 BLACK POWDER IN PELLETS 1.1 1 Y* 0028 BLACK POWDER, COMPRESSED 1.1 1 Y* 0028 GUNPOWDER IN PELLETS 1.1 1 Y* 0028 GUNPOWDER, COMPRESSED 1.1 1 Y* 0029 DETONATORS, NON-ELECTRIC, FOR BLASTING 1.1 1 Y* 0030 DETONATORS, ELECTRIC, FOR BLASTING 1.1 1 Y* 0033 BOMBS, WITH BURSTING CHARGE 1.1 1 Y* 0034 BOMBS, WITH BURSTING CHARGE 1.1 1 Y* 0035 BOMBS, WITH BURSTING CHARGE 1.2 1 Y* 0037 BOMBS, PHOTO-FLASH 1.1 1 Y* 0038 BOMBS, PHOTO-FLASH 1.1 1 Y* 0039 BOMBS, PHOTO-FLASH 1.2 1 Y* 0042 BOOSTERS, WITHOUT DETONATOR 1.1 1 Y* 0043 BURSTERS, EXPLOSIVE 1.1 -

(PON) on 01/12/2018
Uncontrolled when printed Document supersedes GORT3053-1 Iss 8 and comes into force on 01/12/2018 Appendix 1 GORT3053-1 Issue 8.1 December 2018 Appendix 1 LIST OF DANGEROUS GOODS WITH THEIR UNITED NATIONS NUMBER, DANGEROUS GOODS CLASS AND TOPS COMMODITY CODE Contents Page Latest Issue Contents App 1.1 Dec 2018 List of Dangerous Goods with their App 1.2 Dec 2018 United Nations Number, Dangerous Goods Class and TOPS Commodity Code This issue contains changes published in the Periodical Operating Notice (PON) on 01/12/2018 Working Manual for Rail Staff Handling and Carriage of Dangerous Goods Page App1.1 Uncontrolled when printed Document supersedes GORT3053-1 Iss 8 and comes into force on 01/12/2018 Appendix 1 GORT3053-1 Issue 8.1 December 2018 1.1 Classes 2 – 9 This list, applicable to freight train carriage, is for reference purposes only. It must not be used to allocate United Nations numbers, dangerous goods classifications, subsidiary hazards or packing groups. More than one class or packing group is shown against some substances and in these instances the actual class or packing group, which should be declared by the consignor, will depend upon the properties of the actual substances consigned. UN No Dangerous Goods Class Subsidiary Hazard(s) Packing Group Substance TOPS. Commodity Code 1001 Acetylene, dissolved 2.1 550 1002 Air, compressed 2.2 550 1003 Air, refrigerated liquid 2.2 5.1 542 1005 Ammonia, anhydrous 2.3 8 535, 545 1006 Argon, compressed 2.2 550 1008 Boron trifluoride 2.3 8 535, 550 1009 Bromotrifluoromethane (R 13B1) -

Electrochemical Synthesis of Aluminium Hydride for Hydrogen Storage
J. Environ. Nanotechnol. Volume 6, No.1(2017) pp. 34-38 ISSN (Print): 2279-0748 ISSN (Online): 2319-5541 doi:10.13074/jent.2017.03.171224 Electrochemical Synthesis of Aluminium Hydride for Hydrogen Storage G. Saraswathi1, B. Nirmala2*, N. Rajalakshmi3 1,*2Department of Physics, Sri GVG Visalakshi College for Women, Udumalpet, TN, India. 3International Advanced Research Centre for Powder Metallurgy and New Materials (ARCI), Chennai,TN, India. Abstract Electrochemical Synthesis of Aluminium hydride material was gaining interest due to usage in storing hydrogen for automobiles. Biofuel reduces pollution and various cardiac diseases all over the world. The most preferred method was Electrochemical Synthesis. The chemical method degrades the particles in three days. So the Electrochemical Synthesis was the best way to prepare Aluminium Hydride. The chemicals used were Sodium Aluminium Hydride as electrolyte, Tetrahydrofuran (THF) as a solvent and it was toxic. It was performed in Argon atmosphere. Aluminium Hydride (Alane) formed in the form of pore which gets evaporated within few seconds. Keywords: Alane; Argon; Electrochemical; Storage. 1. INTRODUCTION stationary fuel cell systems. For example, size and weight requirements are very important in automotive The DOE is supporting research to demonstrate applications, while not so significant in stationary viable materials for onboard hydrogen storage. applications. The acceptable noise level is lower for Aluminum hydride (alane – AlH3), having a gravimetric stationary applications. Typically noise in the fuel cell capacity of 10 wt% and volumetric capacity of 149 g systems comes from air and fluid handling devices. H2/L and a desorption temperature of ~60 °C to 175 °C Automobile systems are expected to have a very short (depending on particle size and the addition of catalysts) start-up time while the start-up of a stationary system is has the potential to meet the DOE Targets. -

Structure for UNO/IMCO CODES
Structure for UNO/IMCO CODES ------------------------------------------------------------ Field Name Type Width ------------------------------------------------------------ IMCO CODE Alphanumeric 3 IMDG GROUP Alphanumeric 2 UNO CODE Alphanumeric 5 DESCRIPTION Alphanumeric 300 ------------------------------------------------------------ 1.4D 0012 CARTRIDGES SAFETY OTHER THAN BLANK 1.4D 0014 CARTRIDGES SAFETY BLANK 1.4D 0323 CARTRIDGES SAFETY 1.1B 0333 FIRE WORKS(1.1G) 1.2B 0334 FIRE WORKS(1.2G) 1.3B 0335 FIRE WORKS(1.3G) 1.4B 0336 FIRE WORKS(1.4G) 1.4B 0337 FIRE WORKS(1.4S) 2.1B 1001 ACETYLENE, DISSOLVED 2.2C 1002 AIR,COMPRESSED 2.2B 1003 AIR,REFRIGERATED LIQUID 2.2C 1005 AMMONIA,ANHYDROUS LIQUEFIED OR AMMONIA SOLUTION 2.2C 1006 ARGON, COMPRESSED 2.2B 1008 BORON FLUORIDE, BORON TRIFLUORIDE 2.2C 1009 BROMOTRIFLUORO METHANE, R13 B1, TRIFLUORO BROMO METHANE 2.2B 1010 BUTADIENE INHIBITED, DIVINYL,INHIBITED 2.2B 1011 BUTANE OR BUTANE MIXTURE 2.2B 1012 BUTENE,BUTYLENE 2.2C 1013 CARBON DIOXIDE,CARBONIC ANHYDRIDE 2.2C 1014 CARBON DIOXIDE AND OXYGEN MIXTURES, OXYGNE AND CARBON DIOXIDE, MIXTURES 2.2C 1015 CARBON DIOXIDE AND NITROUS OXIDE MIXTURES 2.2B 1016 CARBON MONOXIDE 2.3C 1017 CHLORINE 2.2C 1018 CHLORO DIFLUOROMETHANE, MONOCHLORO DIFLUORO METHANE, R22 2.2C 1020 CHLORO PENTA FLUORO ETHANE, MONOCHLORO PETA FLUORO ETHANE, R115 2.2C 1021 CHLOROTETRA FLUORO ETHANE, MONOCHLORO TETRAFLUORO ETHANE, R124 2.2C 1022 CHLORO TRIFLUORO METHANE, MONOCHLORO TRIFLUORO METHANE, R13, TRIFLUORO CHLORO METHANE 2.2C 1023 COAL GAS 2.2B 1026 CYANOGEN, CYANOGEN, DICYANOGEN -

Aluminium Hydrides and Borohydrides
ALUMINIUM HYDRIDES AND BOROHYDRIDES www.acros.com Contents 1 - Introduction . 1 2 - Synthesis and Properties . 2 3 - Chemistry . 5 3.1 Alkylaluminium compounds as Ziegler-Natta co-catalysts . 5 3.2 Complex aluminium hydrides and borohydrides as reducing agents . 7 3.3 The parent compounds lithium aluminium hydride and sodium borohydride . 8 3.3.1. Lithium aluminium hydride. 8 3.3.2. Sodium borohydride and Sodium borodeuteride . 12 3.4 Tuning of the reactivity by different substituents: Derivatives with different sterical and electronical properties . 16 3.5 Modified borohydrides . 17 3.5.1. Lithium borohydride . 17 3.5.2. Potassium borohydride . 18 3.5.3. Tetraalkylammonium- and tetraalkylphosphonium borohydrides . 18 3.5.4. Calcium borohydride . 19 3.5.5. Sodium cyanoborohydride . 19 3.5.6. Sodium triacetoxyborohydride . 22 3.5.7. Tetramethylammonium triacetoxyborohydride (NEW: AO 39285) . 23 3.6 Alkoholate modified aluminium hydrides . 25 3.6.1. Lithium triethoxyaluminium hydride . 25 3.6.2. Lithium tri-tert-butoxyaluminohydride . 25 3.6.3 Sodium bis(2-methoxyethoxy)aluminiumhydride (“SMEAH”) . 25 3.7 Alkylsubstituted borohydrides . 26 3.7.1. Triethylborohydrides . 26 3.7.2. Tri sec-butylborohydrides and trisamylborohydrides . 29 3.7.3. Lithium trisamylborohydride . 30 3.8. Alkylsubstituted aluminiumhydrides . 30 Index . 36 1 - Introduction The importance of organo-boron and organo-aluminium compounds for science and technology has resulted in three Nobel-prizes for Herbert C. Brown for his work on hydroboration1, for Giulio 1Natta -

Material Safety Data Sheet Sodium Aluminium Hydride MSDS
Material Safety Data Sheet Sodium aluminium hydride MSDS# 10695 Section 1 - Chemical Product and Company Identification MSDS Name: Sodium aluminium hydride Catalog Numbers: AC380110000, AC380110250, AC380111000 Synonyms: Acros Organics BVBA Company Identification: Janssen Pharmaceuticalaan 3a 2440 Geel, Belgium Acros Organics Company Identification: (USA) One Reagent Lane Fair Lawn, NJ 07410 For information in the US, call: 800-ACROS-01 For information in Europe, call: +32 14 57 52 11 Emergency Number, Europe: +32 14 57 52 99 Emergency Number US: 201-796-7100 CHEMTREC Phone Number, US: 800-424-9300 CHEMTREC Phone Number, Europe: 703-527-3887 Section 2 - Composition, Information on Ingredients ---------------------------------------- CAS#: 13770-96-2 Chemical Name: Sodium aluminium hydride %: EINECS#: 237-400-1 ---------------------------------------- Hazard Symbols: F C Risk Phrases: 11 14/15 22 34 Section 3 - Hazards Identification EMERGENCY OVERVIEW Not available Target Organs: Respiratory system, eyes, skin. Potential Health Effects Eye: Causes eye burns. Skin: Causes skin burns. Ingestion: Harmful if swallowed. Causes gastrointestinal tract burns. Inhalation: Causes chemical burns to the respiratory tract. Chronic: Section 4 - First Aid Measures Immediately flush eyes with plenty of water for at least 15 minutes, occasionally lifting the upper and lower Eyes: eyelids. Get medical aid immediately. Get medical aid immediately. Immediately flush skin with plenty of water for at least 15 minutes while Skin: removing contaminated clothing and shoes. Ingestion: Do not induce vomiting. Get medical aid immediately. Get medical aid immediately. Remove from exposure and move to fresh air immediately. If not breathing, Inhalation: give artificial respiration. If breathing is difficult, give oxygen. Notes to Treat symptomatically and supportively. -

Solid State Hydrogen Storage in Alanates and Alanate-Based Compounds: a Review
metals Review Solid State Hydrogen Storage in Alanates and Alanate-Based Compounds: A Review Chiara Milanese 1,* ID , Sebastiano Garroni 2 ID , Fabiana Gennari 3, Amedeo Marini 1, Thomas Klassen 4,5, Martin Dornheim 4 and Claudio Pistidda 4 ID 1 Pavia Hydrogen Lab, C.S.G.I. & Chemistry Department, University of Pavia, Viale Taramelli, 16, 27100 Pavia, Italy; [email protected] 2 International Research Centre in Critical Raw Materials-ICCRAM, University of Burgos, 09001 Burgos, Spain; [email protected] 3 National Council of Scientific and Technological Research (CONICET), Bariloche Atomic Center (National Commission of Atomic Energy) and Balseiro Institute (University of Cuyo) Av. Bustillo 9500, San Carlos de Bariloche, Río Negro 8400, Argentina; [email protected] 4 Institute of Materials Research, Helmholtz-Zentrum Geesthacht, Max-Planck-Straße 1, D-21502 Geesthacht, Germany; [email protected] or [email protected] (T.K.); [email protected] (M.D.); [email protected] (C.P.) 5 Institute of Materials Technology, Helmut Schmidt University, University of the Federal Armed Forces, Holstenhofweg 85, D-22043 Hamburg, Germany * Correspondence: [email protected]; Tel.: +39-0382-987-670 Received: 20 June 2018; Accepted: 18 July 2018; Published: 24 July 2018 Abstract: The safest way to store hydrogen is in solid form, physically entrapped in molecular form in highly porous materials, or chemically bound in atomic form in hydrides. Among the different families of these compounds, alkaline and alkaline earth metals alumino-hydrides (alanates) have been regarded as promising storing media and have been extensively studied since 1997, when Bogdanovic and Schwickardi reported that Ti-doped sodium alanate could be reversibly dehydrogenated under moderate conditions. -

Part 3 Dangerous Goods List and Limited Quantities
PART 3 DANGEROUS GOODS LIST AND LIMITED QUANTITIES EXCEPTIONS - 145 - CHAPTER 3.1 GENERAL 3.1.1 Scope and general provisions 3.1.1.1 The Dangerous Goods List in this Chapter lists the dangerous goods most commonly carried but is not exhaustive. It is intended that the list cover, as far as practicable, all dangerous substances of commercial importance. 3.1.1.2 Where a substance or article is specifically listed by name in the Dangerous Goods List, it shall be transported in accordance with the provisions in the List which are appropriate for that substance or article. A "generic" or "not otherwise specified" entry may be used to permit the transport of substances or articles which do not appear specifically by name in the Dangerous Goods List. Such a substance or article may be transported only after its dangerous properties have been determined. The substance or article shall then be classified according to the class definitions and test criteria and the name in the Dangerous Goods List which most appropriately describes the substance or article shall be used. The classification shall be made by the appropriate competent authority when so required or may otherwise be made by the consignor. Once the class of the substance or article has been so established, all conditions for dispatch and transport, as provided in these Regulations shall be met. Any substance or article having or suspected of having explosive characteristics shall first be considered for inclusion in Class 1. Some collective entries may be of the "generic" or "not otherwise specified" type provided that the regulations contain provisions ensuring safety, both by excluding extremely dangerous goods from normal transport and by covering all subsidiary risks inherent in some goods. -

Safety Data Sheet
PIGAL s.p.a. Revision nr. 6 Dated 5/5/2015 PULITORE PER GITANG - GITANG CLEANER Printed on 03/03/2016 Page n. 1/14 Safety data sheet SECTION 1. Identification of the substance/mixture and of the company/undertaking 1.1. Product identifier Code: F00018 Product name PULITORE PER GITANG - GITANG CLEANER 1.2. Relevant identified uses of the substance or mixture and uses advised against Intended use Cleaner/adhesion promoter for PVC, solvent -based. 1.3. Details of the supplier of the safety data sheet Name PIGAL s.p.a. Full address Via G. Rossa, 2 District and Country 40053 VALSAMOGGIA - Crespellano (BO) ITALIA Tel. +39 051969068 Fax +39 051969353 e-mail address of the competent person responsible for the Safety Data Sheet [email protected]; [email protected] 1.4. Emergency telephone number For urgent inquiries refer to +39 051969068 ore ufficio (8.30 -13; 14 -17.30) 118 (contattare il centro antiveleni più vicino) SECTION 2. Hazards identification. 2.1. Classification of the substance or mixture. The product is classified as hazardous pursuant to the provisions set forth in EC Regulation 1272/2008 (CLP) (and subsequent amendments and supplements). The product thus requires a safety datasheet that complies with the provisions of EC Regulation 1907/2006 and subsequent amendments. Any additional information concerning the risks for health and/or the environment are given in sections 11 and 12 of this sheet. 2.1.1. Regulation 1272/2008 (CLP) and following amendments and adjustments. Hazard classification and indication: Flam. Liq. 2 H225 Carc. 2 H351 Eye Irrit.