Beryllium Chloride Apparatus. Figure M
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Densifying Metal Hydrides with High Temperature and Pressure
3,784,682 United States Patent Office Patented Jan. 8, 1974 feet the true density. That is, by this method only theo- 3,784,682 retical or near theoretical densities can be obtained by DENSIFYING METAL HYDRIDES WITH HIGH making the material quite free from porosity (p. 354). TEMPERATURE AND PRESSURE The true density remains the same. Leonard M. NiebylsM, Birmingham, Mich., assignor to Ethyl Corporation, Richmond, Va. SUMMARY OF THE INVENTION No Drawing. Continuation-in-part of abandoned applica- tion Ser. No. 392,370, Aug. 24, 1964. This application The process of this invention provides a practical Apr. 9,1968, Ser. No. 721,135 method of increasing the true density of hydrides of Int. CI. COlb 6/00, 6/06 metals of Groups II-A, II-B, III-A and III-B of the U.S. CI. 423—645 8 Claims Periodic Table. More specifically, true densities of said 10 metal hydrides may be substantially increased by subject- ing a hydride to superatmospheric pressures at or above ABSTRACT OF THE DISCLOSURE fusion temperatures. When beryllium hydride is subjected A method of increasing the density of a hydride of a to this process, a material having a density of at least metal of Groups II-A, II-B, III-A and III-B of the 0.69 g./cc. is obtained. It may or may not be crystalline. Periodic Table which comprises subjecting a hydride to 15 a pressure of from about 50,000 p.s.i. to about 900,000 DESCRIPTION OF THE PREFERRED p.s.i. at or above the fusion temperature of the hydride; EMBODIMENT i.e., between about 65° C. -

Global Minimum Beryllium Hydride Sheet with Novel Negative Poisson's Ratio: first-Principles Cite This: RSC Adv.,2018,8, 19432 Calculations
RSC Advances View Article Online PAPER View Journal | View Issue Global minimum beryllium hydride sheet with novel negative Poisson's ratio: first-principles Cite this: RSC Adv.,2018,8, 19432 calculations Feng Li, *ab Urs Aeberhard,b Hong Wu,a Man Qiaoc and Yafei Li *c As one of the most prominent metal-hydrides, beryllium hydride has received much attention over the past several decades, since 1978, and is considered as an important hydrogen storage material. By reducing the dimensionality from 3 to 2, the beryllium hydride monolayer is isoelectronic with graphene; thus the existence of its two-dimensional (2D) form is theoretically feasible and experimentally expected. However, little is known about its 2D form. In this work, by a global minimum search with the particle swarm optimization method via density functional theory computations, we predicted two new stable structures for the beryllium hydride sheets, named a–BeH2 and b–BeH2 monolayers. Both structures have more favorable thermodynamic stability than the recently reported planar square form (Nanoscale, Creative Commons Attribution-NonCommercial 3.0 Unported Licence. 2017, 9, 8740), due to the forming of multicenter delocalized Be–H bonds. Utilizing the recently developed SSAdNDP method, we revealed that three-center-two-electron (3c–2e) delocalized Be–H bonds are formed in the a–BeH2 monolayer, while for the b–BeH2 monolayer, novel four-center-two- electron (4c–2e) delocalized bonds are observed in the 2D system for the first time. These unique multicenter chemical bonds endow both a– and b–BeH2 with high structural stabilities, which are further confirmed by the absence of imaginary modes in their phonon spectra, the favorable formation energies comparable to bulk and cluster beryllium hydride, and the high mechanical strength. -

United States Patent 19 11 Patent Number: 5,418,243 Angerbauer Et Al
USOO5418243A United States Patent 19 11 Patent Number: 5,418,243 Angerbauer et al. 45 Date of Patent: May 23, 1995 54 SUBSTITUTED 4-PHENYL-PYRIDONES 4,215,126 7/1980. Durant et al........................ 514/345 AND 4-PHENYL-3-ALKOXYPYRIDNES 4,684,477 8/1987 Sugimori et al. ... 546/290 4,916,239 4/1990 Treiber ................. ... 549/292 75 Inventors: Rolf Angerbauer; Peter Fey; Walter 4,988,711 1/1991 Angerbauer et al. ... 514/326 Hibsch, all of Wuppertal; Thomas 5,032,602 7/1991 Fey et al. ................. ... 54/345 Philipps, Cologne; Hilmar Bischoff, 5,064,841 11/1991 Angerbauer et al. ... 514/336 Wuppertal; Hans-Peter Krause, 5,138,090 8/1992 Fey et al. .............................. 560/59 Schwelm; Jörg Peterson-von Gehr, Bochum; Delf Schmidt, Wuppertal, FOREIGN PATENT DOCUMENTS all of Germany 0373423 6/1990 European Pat. Off. 73 Assignee: Bayer Aktiengesellschaft, OTHER PUBLICATIONS Leverkusen, Germany J. Med. Chem., 1990, vol. 33, pp. 52-60; “Synthesis and 21) Appl. No.: 166,775 Biological Activity of New HGM-CoA Reductase 22 Filed: Dec. 14, 1993 Inhibitors ... ', G. Beck. Primary Examiner-C. Warren Ivy (30) Foreign Application Priority Data Assistant Examiner-A. A. Owens Dec. 21, 1992 DE Germany ........................ 4243 278.2 Attorney, Agent, or Firm-Sprung, Horn, Kramer & Jun. 28, 1993 DE Germany ........................ 43 21421.5 Woods 511 Int. Cl...................... A61K 31/44; C07D 213/64 57 ABSTRACT 52 U.S. C. ...................................... 514/345; 546/24; 546/290; 514/89 Substituted 4-phenyl-pyridones and 4-phenyl-2-alkox 58 Field of Search .................. 546/290, 24; 514/345, ypyridines are prepared by reducing corresponding 514/89 4-phenyl-pyridone and 4-phenyl-2-alkoxypyridine de rivatives. -
Chapter 13.Pptx
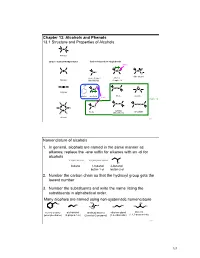
Chapter 13: Alcohols and Phenols 13.1 Structure and Properties of Alcohols C C Alkanes Carbon - Carbon Multiple Bonds Carbon-heteroatom single bonds basic O C C C N C N C X O nitro alkane X= F, Cl, Br, I amines Alkenes Alkyl Halide Chapter 23 OH C C H O C O C C O C C Alkynes phenol alcohols ethers epoxide acidic Chapter 14 H H H C S C C C C S S C C S C C H C C sulfides thiols disulfide H H (thioethers) Arenes 253 Nomenclature of alcohols 1. In general, alcohols are named in the same manner as alkanes; replace the -ane suffix for alkanes with an -ol for alcohols CH3CH2CH2CH3 CH3CH2CH2CH2OH OH butane 1-butanol 2-butanol butan-1-ol butan-2-ol 2. Number the carbon chain so that the hydroxyl group gets the lowest number 3. Number the substituents and write the name listing the substituents in alphabetical order. Many alcohols are named using non-systematic nomenclature H C OH 3 OH OH C OH OH HO OH H3C HO H3C benzyl alcohol allyl alcohol tert-butyl alcohol ethylene glycol glycerol (phenylmethanol) (2-propen-1-ol) (2-methyl-2-propanol) (1,2-ethanediol) (1,2,3-propanetriol) 254 127 Alcohols are classified according to the H R C OH C OH H H degree of substitution of the carbon bearing H H 1° carbon the -OH group methanol primary alcohol primary (1°) : one alkyl substituent R R C OH C OH R R secondary (2°) : two alkyl substituents H R 2° carbon 3° carbon tertiary (3°) : three alkyl substituents secondary alcohol tertiary alcohol Physical properties of alcohols – the C-OH bond of alcohols has a significant dipole moment. -

Radical Approaches to Alangium and Mitragyna Alkaloids
Radical Approaches to Alangium and Mitragyna Alkaloids A Thesis Submitted for a PhD University of York Department of Chemistry 2010 Matthew James Palframan Abstract The work presented in this thesis has focused on the development of novel and concise syntheses of Alangium and Mitragyna alkaloids, and especial approaches towards (±)-protoemetinol (a), which is a key precursor of a range of Alangium alkaloids such as psychotrine (b) and deoxytubulosine (c). The approaches include the use of a key radical cyclisation to form the tri-cyclic core. O O O N N N O O O H H H H H H O N NH N Protoemetinol OH HO a Psychotrine Deoxytubulosine b c Chapter 1 gives a general overview of radical chemistry and it focuses on the application of radical intermolecular and intramolecular reactions in synthesis. Consideration is given to the mediator of radical reactions from the classic organotin reagents, to more recently developed alternative hydrides. An overview of previous synthetic approaches to a range of Alangium and Mitragyna alkaloids is then explored. Chapter 2 follows on from previous work within our group, involving the use of phosphorus hydride radical addition reactions, to alkenes or dienes, followed by a subsequent Horner-Wadsworth-Emmons reaction. It was expected that the tri-cyclic core of the Alangium alkaloids could be prepared by cyclisation of a 1,7-diene, using a phosphorus hydride to afford the phosphonate or phosphonothioate, however this approach was unsuccessful and it highlighted some limitations of the methodology. Chapter 3 explores the radical and ionic chemistry of a range of silanes. -

Reductions and Reducing Agents
REDUCTIONS AND REDUCING AGENTS 1 Reductions and Reducing Agents • Basic definition of reduction: Addition of hydrogen or removal of oxygen • Addition of electrons 9:45 AM 2 Reducible Functional Groups 9:45 AM 3 Categories of Common Reducing Agents 9:45 AM 4 Relative Reactivity of Nucleophiles at the Reducible Functional Groups In the absence of any secondary interactions, the carbonyl compounds exhibit the following order of reactivity at the carbonyl This order may however be reversed in the presence of unique secondary interactions inherent in the molecule; interactions that may 9:45 AM be activated by some property of the reacting partner 5 Common Reducing Agents (Borohydrides) Reduction of Amides to Amines 9:45 AM 6 Common Reducing Agents (Borohydrides) Reduction of Carboxylic Acids to Primary Alcohols O 3 R CO2H + BH3 R O B + 3 H 3 2 Acyloxyborane 9:45 AM 7 Common Reducing Agents (Sodium Borohydride) The reductions with NaBH4 are commonly carried out in EtOH (Serving as a protic solvent) Note that nucleophilic attack occurs from the least hindered face of the 8 carbonyl Common Reducing Agents (Lithium Borohydride) The reductions with LiBH4 are commonly carried out in THF or ether Note that nucleophilic attack occurs from the least hindered face of the 9:45 AM 9 carbonyl. Common Reducing Agents (Borohydrides) The Influence of Metal Cations on Reactivity As a result of the differences in reactivity between sodium borohydride and lithium borohydride, chemoselectivity of reduction can be achieved by a judicious choice of reducing agent. 9:45 AM 10 Common Reducing Agents (Sodium Cyanoborohydride) 9:45 AM 11 Common Reducing Agents (Reductive Amination with Sodium Cyanoborohydride) 9:45 AM 12 Lithium Aluminium Hydride Lithium aluminiumhydride reacts the same way as lithium borohydride. -

S-Block Amidoboranes: Syntheses, Structures, Reactivity and Applications Cite This: Chem
Chem Soc Rev View Article Online REVIEW ARTICLE View Journal | View Issue s-Block amidoboranes: syntheses, structures, reactivity and applications Cite this: Chem. Soc. Rev., 2016, 45, 1112 Tom E. Stennett and Sjoerd Harder* Metal amidoborane compounds of the alkali- and alkaline earth metals have in recent years found applications in diverse disciplines, notably as hydrogen storage materials, as reagents for the reduction of organic functional groups and as catalysts and intermediates in dehydrocoupling reactions. These functions are connected by the organometallic chemistry of the MNR2BH3 group.† This review focusses on central aspects of the s-block amidoborane compounds – their syntheses, structures and reactivity. Well-defined amidoborane complexes of group 2 metals are now available by a variety of solution-phase routes, which has allowed a more detailed analysis of this functional group, which was previously largely confined to solid-state materials chemistry. Structures obtained from X-ray crystallography have begun to provide increased understanding of the fundamental steps of key processes, including amine–borane Creative Commons Attribution 3.0 Unported Licence. dehydrocoupling and hydrogen release from primary and secondary amidoboranes. We review structural parameters and reactivity to rationalise the effects of the metal, nitrogen substituents and supporting Received 10th July 2015 ligands on catalytic performance and dehydrogenative decomposition routes. Mechanistic features of DOI: 10.1039/c5cs00544b key processes involving amidoborane compounds as starting materials or intermediates are discussed, alongside emerging applications such as the use of group 1 metal amidoboranes in synthesis. Finally, the www.rsc.org/chemsocrev future prospects of this vibrant branch of main group chemistry are evaluated. -

(12) Patent Application Publication (10) Pub. No.: US 2005/0044778A1 Orr (43) Pub
US 20050044778A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2005/0044778A1 Orr (43) Pub. Date: Mar. 3, 2005 (54) FUEL COMPOSITIONS EMPLOYING Publication Classification CATALYST COMBUSTION STRUCTURE (51) Int. CI.' ........ C10L 1/28; C1OL 1/24; C1OL 1/18; (76) Inventor: William C. Orr, Denver, CO (US) C1OL 1/12; C1OL 1/26 Correspondence Address: (52) U.S. Cl. ................. 44/320; 44/435; 44/378; 44/388; HOGAN & HARTSON LLP 44/385; 44/444; 44/443 ONE TABOR CENTER, SUITE 1500 1200 SEVENTEENTH ST DENVER, CO 80202 (US) (57) ABSTRACT (21) Appl. No.: 10/722,127 Metallic vapor phase fuel compositions relating to a broad (22) Filed: Nov. 24, 2003 Spectrum of pollution reducing, improved combustion per Related U.S. Application Data formance, and enhanced Stability fuel compositions for use in jet, aviation, turbine, diesel, gasoline, and other combus (63) Continuation-in-part of application No. 08/986,891, tion applications include co-combustion agents preferably filed on Dec. 8, 1997, now Pat. No. 6,652,608. including trimethoxymethylsilane. Patent Application Publication Mar. 3, 2005 US 2005/0044778A1 FIGURE 1 CALCULATING BUNSEN BURNER LAMINAR FLAME VELOCITY (LFV) OR BURNING VELOCITY (BV) CONVENTIONAL FLAME LUMINOUS FLAME Method For Calculating Bunsen Burner Laminar Flame Velocity (LHV) or Burning Velocity Requires Inside Laminar Cone Angle (0) and The Gas Velocity (Vg). LFV = A, SIN 2 x VG US 2005/0044778A1 Mar. 3, 2005 FUEL COMPOSITIONS EMPLOYING CATALYST Chart of Elements (CAS version), and mixture, wherein said COMBUSTION STRUCTURE element or derivative compound, is combustible, and option 0001) The present invention is a CIP of my U.S. -

Synthesis, Characterization and Polymerization of Ethylene Using a Novel Soluble Magnesium-Titanium Catalyst*
Synthesis, characterization and polymerization of ethylene using a novel soluble magnesium-titanium catalyst* Ganugupati Satyanarayana and Swaminathan Sivaramt Division of Polymer Chemistry, National Chemical Laboratory, Pune 411008, India (Received 4 January 1993; revised 28 May 1993) A l:2 complex of MgCI2 and tetrahydrofuran (THF) was prepared by a simple Grignard decomposition reaction using Mg metal and 1,2-dichloroethane in THF as diluent. The MgC12.2THF complex formed a homogeneous solution with titanium-n-butoxide in xylene at 60°C. The soluble Mg-Ti complex in xylene polymerized ethylene under homogeneous conditions in the presence of organoaluminium compounds. The kinetic behaviour of the polymerization reaction was studied. It was observed that the presence of MgCI2 transformed the Ti centre from a dimerization to a polymerization catalyst. (Keywords: magnesium chloride; titanium-n-butoxide; magnesium-titanium catalyst) INTRODUCTION a Grignard decomposition reaction forms a soluble complex with titanium-n-butoxide, which in conjunction Solid catalysts based on anhydrous MgCI z and Ti halides with organoaluminium compound initiates ethylene have attracted considerable attention during the past polymerization under homogeneous conditions in xylene decade as components of high efficiency ethylene and as the solvent 11. This paper reports our results on the propylene polymerization catalysts 1. The role of MgC12 synthesis, characterization and polymerization of ethylene in these catalysts has been widely discussed in the using this novel soluble Mg-Ti catalyst. literature 2. There are, however, only a few reports of truly soluble Mg-Ti catalysts for olefin polymerization. Soga et al. prepared a soluble metal halide-2-ethylhexanol complex which, in conjunction with titanium-n-butoxide EXPERIMENTAL and diethylaluminium chloride, polymerized propylene 3. -

Global Minimum Beryllium Hydride Sheet with Novel Negative Poisson's Ratio: first-Principles Cite This: RSC Adv.,2018,8, 19432 Calculations
RSC Advances View Article Online PAPER View Journal | View Issue Global minimum beryllium hydride sheet with novel negative Poisson's ratio: first-principles Cite this: RSC Adv.,2018,8, 19432 calculations Feng Li, *ab Urs Aeberhard,b Hong Wu,a Man Qiaoc and Yafei Li *c As one of the most prominent metal-hydrides, beryllium hydride has received much attention over the past several decades, since 1978, and is considered as an important hydrogen storage material. By reducing the dimensionality from 3 to 2, the beryllium hydride monolayer is isoelectronic with graphene; thus the existence of its two-dimensional (2D) form is theoretically feasible and experimentally expected. However, little is known about its 2D form. In this work, by a global minimum search with the particle swarm optimization method via density functional theory computations, we predicted two new stable structures for the beryllium hydride sheets, named a–BeH2 and b–BeH2 monolayers. Both structures have more favorable thermodynamic stability than the recently reported planar square form (Nanoscale, Creative Commons Attribution-NonCommercial 3.0 Unported Licence. 2017, 9, 8740), due to the forming of multicenter delocalized Be–H bonds. Utilizing the recently developed SSAdNDP method, we revealed that three-center-two-electron (3c–2e) delocalized Be–H bonds are formed in the a–BeH2 monolayer, while for the b–BeH2 monolayer, novel four-center-two- electron (4c–2e) delocalized bonds are observed in the 2D system for the first time. These unique multicenter chemical bonds endow both a– and b–BeH2 with high structural stabilities, which are further confirmed by the absence of imaginary modes in their phonon spectra, the favorable formation energies comparable to bulk and cluster beryllium hydride, and the high mechanical strength. -

Doped Acetylene Polymer and Process for Production Thereof
European Patent Office © Publication number: 0 022 271 A1 Office europeen des brevets © EUROPEAN PATENT APPLICATION © Application number: 80103857.1 © Int. CI.3: C 08 L 49/00 C 08 J 3/20 © Date of filing: 07.07.80 //C08K3/10, C08K5/08, C08K5/09, H01B1/00 © Priority: 10.07.79 JP 86402/79 © Applicant: JAPAN SYNTHETIC RUBBER CO., LTD. 10.07.79 JP 8640379 11-24, Tsukiji-2-chome Chuo-ku 28.08.79 JP 108641/79 Tokyo(JP) © Inventor: Matsumura, Yoshio © Date of publication of application: 14-30, Tsukimino-8-chome 14.01.81 Bulletin 81/2 Yamato-shi(JP) © Designated Contracting States: © Inventor: Nozue, Ikuo DE FR GB NL 29, Aobadai-2-chome Midori-ku, Yokohama(JP) © Inventor: Ukachi, Takashi 29, Aobadai-2-chome Midori-ku, Yokohama(JP) © Representative: Beetz, sen., Richard, Dipl. -Ing. Patentanwalte Dipl. -Ing. R. Beetz sen. Dipl.-lng. K. Lamprecht;Dr. Ing. R. Beetz jr. et al, Rechtsanwalt Dipl.-Phys. Dr. jur. U. Heidrich Dr.-lng. W. Timpe; Dipl.-lng. J. Siegfried Dipl.-Chem. Dr.rer.nat.W. Schmitt-Fumian Steinsdorfstrasse 10 D-8000 Munchen 22(DE) © Doped acetylene polymer and process for production thereof. (57) Doped acetylene polymers are produced by immersing an acetylene polymer under an inert gas atmosphere in an organic solvent solution of a dopant selected from the group consisting of a platinum group metal complex, a carbonium salt, an oxonium salt and a parabenzoquinone derivative. According to this process, a doped acetylene polymer having any desired electrical conductivity can be produced and the doped acetylene polymer thus obtained has excellent proper- ties as an organic semiconductor material for solar batteries, various sensors, etc. -

Flexible Solar Cells
Flexible Solar Cells A Major Qualifying Project Submitted to the Faculty Of Worcester Polytechnic Institute In Partial Fulfillment of the requirements for the Degree in Bachelor of Science In Mechanical Engineering By Francis LaRovere Edward Peglow Michael McMahon Project Advisor Professor Pratap Rao, Advisor This report represents work of WPI undergraduate students submitted to the faculty as evidence of a degree requirement. WPI routinely publishes these reports on its w ebsite without editorial or peer review. For more information about the projects program at WPI, see http://www.wpi.edu/Academics/Projects. 1 Table of Contents Table of Figures ............................................................................................................................................. 5 Table of Tables .............................................................................................................................................. 6 Acknowledgments ......................................................................................................................................... 7 Abstract ......................................................................................................................................................... 8 1. Introduction .............................................................................................................................................. 9 2. Scope ......................................................................................................................................................