Solid State Hydrogen Storage in Alanates and Alanate-Based Compounds: a Review
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

United States Patent 19 11 Patent Number: 5,418,243 Angerbauer Et Al
USOO5418243A United States Patent 19 11 Patent Number: 5,418,243 Angerbauer et al. 45 Date of Patent: May 23, 1995 54 SUBSTITUTED 4-PHENYL-PYRIDONES 4,215,126 7/1980. Durant et al........................ 514/345 AND 4-PHENYL-3-ALKOXYPYRIDNES 4,684,477 8/1987 Sugimori et al. ... 546/290 4,916,239 4/1990 Treiber ................. ... 549/292 75 Inventors: Rolf Angerbauer; Peter Fey; Walter 4,988,711 1/1991 Angerbauer et al. ... 514/326 Hibsch, all of Wuppertal; Thomas 5,032,602 7/1991 Fey et al. ................. ... 54/345 Philipps, Cologne; Hilmar Bischoff, 5,064,841 11/1991 Angerbauer et al. ... 514/336 Wuppertal; Hans-Peter Krause, 5,138,090 8/1992 Fey et al. .............................. 560/59 Schwelm; Jörg Peterson-von Gehr, Bochum; Delf Schmidt, Wuppertal, FOREIGN PATENT DOCUMENTS all of Germany 0373423 6/1990 European Pat. Off. 73 Assignee: Bayer Aktiengesellschaft, OTHER PUBLICATIONS Leverkusen, Germany J. Med. Chem., 1990, vol. 33, pp. 52-60; “Synthesis and 21) Appl. No.: 166,775 Biological Activity of New HGM-CoA Reductase 22 Filed: Dec. 14, 1993 Inhibitors ... ', G. Beck. Primary Examiner-C. Warren Ivy (30) Foreign Application Priority Data Assistant Examiner-A. A. Owens Dec. 21, 1992 DE Germany ........................ 4243 278.2 Attorney, Agent, or Firm-Sprung, Horn, Kramer & Jun. 28, 1993 DE Germany ........................ 43 21421.5 Woods 511 Int. Cl...................... A61K 31/44; C07D 213/64 57 ABSTRACT 52 U.S. C. ...................................... 514/345; 546/24; 546/290; 514/89 Substituted 4-phenyl-pyridones and 4-phenyl-2-alkox 58 Field of Search .................. 546/290, 24; 514/345, ypyridines are prepared by reducing corresponding 514/89 4-phenyl-pyridone and 4-phenyl-2-alkoxypyridine de rivatives. -

CNSC Research Report 2016-17
The Science of Safety: CNSC Research Report 2016–17 © Canadian Nuclear Safety Commission (CNSC) 2018 Cat. No. CC171-24E-PDF ISSN 2369-4351 Extracts from this document may be reproduced for individual use without permission provided the source is fully acknowledged. However, reproduction in whole or in part for purposes of resale or redistribution requires prior written permission from the Canadian Nuclear Safety Commission. Également publié en français sous le titre : La science de la sûreté : Rapport de recherche de la CCSN 2016-2017 Document availability This document can be viewed on the CNSC website. To request a copy of the document in English or French, please contact: Canadian Nuclear Safety Commission 280 Slater Street P.O. Box 1046, Station B Ottawa, Ontario K1P 5S9 CANADA Tel.: 613-995-5894 or 1-800-668-5284 (in Canada only) Facsimile: 613-995-5086 Email: [email protected] Website: nuclearsafety.gc.ca Facebook: facebook.com/CanadianNuclearSafetyCommission YouTube: youtube.com/cnscccsn Twitter: @CNSC_CCSN Publishing History June 2018 Edition 1.0 Table of contents Message from the President .......................................................................................................................... 1 Introduction ................................................................................................................................................... 2 Ensuring the safety of nuclear power plants ................................................................................................. 6 Protecting -

Source of Atomic Hydrogen for Ion Trap Experiments: Review and Basic Properties
WDS'15 Proceedings of Contributed Papers — Physics, 155–161, 2015. ISBN 978-80-7378-311-2 © MATFYZPRESS Source of Atomic Hydrogen for Ion Trap Experiments: Review and Basic Properties A. Kovalenko, Š Roučka, S. Rednyk, T. D. Tran, D. Mulin, R. Plašil, J. Glosík Charles University in Prague, Faculty of Mathematics and Physics, Prague, Czech Republic. Abstract. The H-atom source was used to produce atomic hydrogen for study of ion-molecule reactions relevant to astrochemistry at low temperatures. H atoms were cooled and formed into an effusive beam, passing through a 22-pole ion trap. Here we present the basic operating principles of the H-atom source and give a review of this apparatus with some calculations of vacuum conditions and parameters of the produced H-atom beam. Introduction Hydrogen is the most abundant element in the Universe [Field et al., 1966]. It is the main component of stars, giant planets and interstellar clouds. Reactions with atomic or molecular hydrogen are important for understanding the processes which occur in the interstellar medium and during the formation of the new stars. There are three isotopes of hydrogen: protium, deuterium and tritium. The reactions of ions with H atoms have to be studied for better understanding of formation and destruction of more complex ions and molecules observed in interstellar medium. There are regions in space called H I and H II regions where hydrogen is mostly neutral in H I regions, rather than ionized or molecular in the H II regions. The atomic hydrogen plays fundamental role in many astrophysical contexts especially in formation H2 molecules [Habart et al., 2005; Sternberg et al., 2014]. -

Understanding Variation in Partition Coefficient, Kd, Values: Volume II
United States Office of Air and Radiation EPA 402-R-99-004B Environmental Protection August 1999 Agency UNDERSTANDING VARIATION IN PARTITION COEFFICIENT, Kd, VALUES Volume II: Review of Geochemistry and Available Kd Values for Cadmium, Cesium, Chromium, Lead, Plutonium, Radon, Strontium, Thorium, Tritium (3H), and Uranium UNDERSTANDING VARIATION IN PARTITION COEFFICIENT, Kd, VALUES Volume II: Review of Geochemistry and Available Kd Values for Cadmium, Cesium, Chromium, Lead, Plutonium, Radon, Strontium, Thorium, Tritium (3H), and Uranium August 1999 A Cooperative Effort By: Office of Radiation and Indoor Air Office of Solid Waste and Emergency Response U.S. Environmental Protection Agency Washington, DC 20460 Office of Environmental Restoration U.S. Department of Energy Washington, DC 20585 NOTICE The following two-volume report is intended solely as guidance to EPA and other environmental professionals. This document does not constitute rulemaking by the Agency, and cannot be relied on to create a substantive or procedural right enforceable by any party in litigation with the United States. EPA may take action that is at variance with the information, policies, and procedures in this document and may change them at any time without public notice. Reference herein to any specific commercial products, process, or service by trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government. ii FOREWORD Understanding the long-term behavior of contaminants in the subsurface is becoming increasingly more important as the nation addresses groundwater contamination. Groundwater contamination is a national concern as about 50 percent of the United States population receives its drinking water from groundwater. -
Chapter 13.Pptx
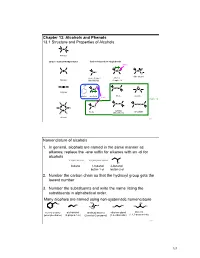
Chapter 13: Alcohols and Phenols 13.1 Structure and Properties of Alcohols C C Alkanes Carbon - Carbon Multiple Bonds Carbon-heteroatom single bonds basic O C C C N C N C X O nitro alkane X= F, Cl, Br, I amines Alkenes Alkyl Halide Chapter 23 OH C C H O C O C C O C C Alkynes phenol alcohols ethers epoxide acidic Chapter 14 H H H C S C C C C S S C C S C C H C C sulfides thiols disulfide H H (thioethers) Arenes 253 Nomenclature of alcohols 1. In general, alcohols are named in the same manner as alkanes; replace the -ane suffix for alkanes with an -ol for alcohols CH3CH2CH2CH3 CH3CH2CH2CH2OH OH butane 1-butanol 2-butanol butan-1-ol butan-2-ol 2. Number the carbon chain so that the hydroxyl group gets the lowest number 3. Number the substituents and write the name listing the substituents in alphabetical order. Many alcohols are named using non-systematic nomenclature H C OH 3 OH OH C OH OH HO OH H3C HO H3C benzyl alcohol allyl alcohol tert-butyl alcohol ethylene glycol glycerol (phenylmethanol) (2-propen-1-ol) (2-methyl-2-propanol) (1,2-ethanediol) (1,2,3-propanetriol) 254 127 Alcohols are classified according to the H R C OH C OH H H degree of substitution of the carbon bearing H H 1° carbon the -OH group methanol primary alcohol primary (1°) : one alkyl substituent R R C OH C OH R R secondary (2°) : two alkyl substituents H R 2° carbon 3° carbon tertiary (3°) : three alkyl substituents secondary alcohol tertiary alcohol Physical properties of alcohols – the C-OH bond of alcohols has a significant dipole moment. -

Crystal Chemistry of Perovskite-Type Hydride Namgh3: Implications for Hydrogen Storage
Chem. Mater. 2008, 20, 2335–2342 2335 Crystal Chemistry of Perovskite-Type Hydride NaMgH3: Implications for Hydrogen Storage Hui Wu,*,†,‡ Wei Zhou,†,‡ Terrence J. Udovic,† John J. Rush,†,‡ and Taner Yildirim†,§ NIST Center for Neutron Research, National Institute of Standards and Technology, 100 Bureau DriVe, MS 6102, Gaithersburg, Maryland 20899-6102, Department of Materials Science and Engineering, UniVersity of Maryland, College Park, Maryland 20742-2115, and Department of Materials Science and Engineering, UniVersity of PennsylVania, 3231 Walnut Street, Philadelphia, PennsylVania 19104-6272 ReceiVed NoVember 26, 2007. ReVised Manuscript ReceiVed January 11, 2008 The crystal structure, lattice dynamics, and local metal-hydrogen bonding of the perovskite hydride NaMgH3 were investigated using combined neutron powder diffraction, neutron vibrational spectroscopy, and first-principles calculations. NaMgH3 crystallizes in the orthorhombic GdFeO3-type perovskite structure (Pnma) with a-b+a- octahedral tilting in the temperature range of 4 to 370 K. In contrast with previous structure studies, the refined Mg-H lengths and H-Mg-H angles indicate that the MgH6 octahedra maintain a near ideal configuration, which is corroborated by bond valence methods and our DFT calculations, and is consistent with perovskite oxides with similar tolerance factor values. The temperature dependences of the lattice distortion, octahedral tilting angle, and atomic displacement of H are also consistent with the recently observed high H mobility at elevated temperature. The stability and dynamics of NaMgH3 are discussed and rationalized in terms of the details of our observed perovskite structure. Further experiments reveal that its perovskite crystal structure and associated rapid hydrogen motion can be used to improve the slow hydrogenation kinetics of some strongly bound light-metal-hydride systems such as MgH2 and possibly to design new alloy hydrides with desirable hydrogen-storage properties. -

Radical Approaches to Alangium and Mitragyna Alkaloids
Radical Approaches to Alangium and Mitragyna Alkaloids A Thesis Submitted for a PhD University of York Department of Chemistry 2010 Matthew James Palframan Abstract The work presented in this thesis has focused on the development of novel and concise syntheses of Alangium and Mitragyna alkaloids, and especial approaches towards (±)-protoemetinol (a), which is a key precursor of a range of Alangium alkaloids such as psychotrine (b) and deoxytubulosine (c). The approaches include the use of a key radical cyclisation to form the tri-cyclic core. O O O N N N O O O H H H H H H O N NH N Protoemetinol OH HO a Psychotrine Deoxytubulosine b c Chapter 1 gives a general overview of radical chemistry and it focuses on the application of radical intermolecular and intramolecular reactions in synthesis. Consideration is given to the mediator of radical reactions from the classic organotin reagents, to more recently developed alternative hydrides. An overview of previous synthetic approaches to a range of Alangium and Mitragyna alkaloids is then explored. Chapter 2 follows on from previous work within our group, involving the use of phosphorus hydride radical addition reactions, to alkenes or dienes, followed by a subsequent Horner-Wadsworth-Emmons reaction. It was expected that the tri-cyclic core of the Alangium alkaloids could be prepared by cyclisation of a 1,7-diene, using a phosphorus hydride to afford the phosphonate or phosphonothioate, however this approach was unsuccessful and it highlighted some limitations of the methodology. Chapter 3 explores the radical and ionic chemistry of a range of silanes. -

Reductions and Reducing Agents
REDUCTIONS AND REDUCING AGENTS 1 Reductions and Reducing Agents • Basic definition of reduction: Addition of hydrogen or removal of oxygen • Addition of electrons 9:45 AM 2 Reducible Functional Groups 9:45 AM 3 Categories of Common Reducing Agents 9:45 AM 4 Relative Reactivity of Nucleophiles at the Reducible Functional Groups In the absence of any secondary interactions, the carbonyl compounds exhibit the following order of reactivity at the carbonyl This order may however be reversed in the presence of unique secondary interactions inherent in the molecule; interactions that may 9:45 AM be activated by some property of the reacting partner 5 Common Reducing Agents (Borohydrides) Reduction of Amides to Amines 9:45 AM 6 Common Reducing Agents (Borohydrides) Reduction of Carboxylic Acids to Primary Alcohols O 3 R CO2H + BH3 R O B + 3 H 3 2 Acyloxyborane 9:45 AM 7 Common Reducing Agents (Sodium Borohydride) The reductions with NaBH4 are commonly carried out in EtOH (Serving as a protic solvent) Note that nucleophilic attack occurs from the least hindered face of the 8 carbonyl Common Reducing Agents (Lithium Borohydride) The reductions with LiBH4 are commonly carried out in THF or ether Note that nucleophilic attack occurs from the least hindered face of the 9:45 AM 9 carbonyl. Common Reducing Agents (Borohydrides) The Influence of Metal Cations on Reactivity As a result of the differences in reactivity between sodium borohydride and lithium borohydride, chemoselectivity of reduction can be achieved by a judicious choice of reducing agent. 9:45 AM 10 Common Reducing Agents (Sodium Cyanoborohydride) 9:45 AM 11 Common Reducing Agents (Reductive Amination with Sodium Cyanoborohydride) 9:45 AM 12 Lithium Aluminium Hydride Lithium aluminiumhydride reacts the same way as lithium borohydride. -

Modeling Gas Absorption
Project Number: WMC 4028 Modeling Gas Absorption A Major Qualifying Project Report submitted to the Faculty of the WORCESTER POLYTECHNIC INSTITUTE in partial fulfillment of the requirements for the Degree of Bachelor of Science by _______________________________________ Yaminah Z. Jackson Date: April 24, 2008 Approved: _________________________________________ Professor William M. Clark, Project Advisor ABSTRACT This project sought to analyze the gas absorption process as an efficient way in which to remove pollutants, such as carbon dioxide from gas streams. The designed absorption lab for CM 4402 was used to collect data based on the change in composition throughout the column. The recorded and necessary calculated values were then used to create a simulation model using COMSOL Multiphysics, as a supplemental learning tool for students in CM 4402. 2 TABLE OF CONTENTS ABSTRACT ................................................................................................................................................................. 2 TABLE OF CONTENTS ............................................................................................................................................ 3 INTRODUCTION ....................................................................................................................................................... 5 ANTHROPOGENIC SOURCES ..................................................................................................................................... 5 FINDING A SOLUTION -

Carbon Dioxide Capture by Chemical Absorption: a Solvent Comparison Study
Carbon Dioxide Capture by Chemical Absorption: A Solvent Comparison Study by Anusha Kothandaraman B. Chem. Eng. Institute of Chemical Technology, University of Mumbai, 2005 M.S. Chemical Engineering Practice Massachusetts Institute of Technology, 2006 SUBMITTED TO THE DEPARTMENT OF CHEMICAL ENGINEERING IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY IN CHEMICAL ENGINEERING PRACTICE AT THE MASSACHUSETTS INSTITUTE OF TECHNOLOGY JUNE 2010 © 2010 Massachusetts Institute of Technology All rights reserved. Signature of Author……………………………………………………………………………………… Department of Chemical Engineering May 20, 2010 Certified by……………………………………………………….………………………………… Gregory J. McRae Hoyt C. Hottel Professor of Chemical Engineering Thesis Supervisor Accepted by……………………………………………………………………………….................... William M. Deen Carbon P. Dubbs Professor of Chemical Engineering Chairman, Committee for Graduate Students 1 2 Carbon Dioxide Capture by Chemical Absorption: A Solvent Comparison Study by Anusha Kothandaraman Submitted to the Department of Chemical Engineering on May 20, 2010 in partial fulfillment of the requirements of the Degree of Doctor of Philosophy in Chemical Engineering Practice Abstract In the light of increasing fears about climate change, greenhouse gas mitigation technologies have assumed growing importance. In the United States, energy related CO2 emissions accounted for 98% of the total emissions in 2007 with electricity generation accounting for 40% of the total1. Carbon capture and sequestration (CCS) is one of the options that can enable the utilization of fossil fuels with lower CO2 emissions. Of the different technologies for CO2 capture, capture of CO2 by chemical absorption is the technology that is closest to commercialization. While a number of different solvents for use in chemical absorption of CO2 have been proposed, a systematic comparison of performance of different solvents has not been performed and claims on the performance of different solvents vary widely. -

Lithium Hydride Powered PEM Fuel Cells for Long-Duration Small Mobile Robotic Missions
Lithium Hydride Powered PEM Fuel Cells for Long-Duration Small Mobile Robotic Missions Jekanthan Thangavelautham, Daniel Strawser, Mei Yi Cheung Steven Dubowsky Abstract— This paper reports on a study to develop power supplies for small mobile robots performing long duration missions. It investigates the use of fuel cells to achieve this objective, and in particular Proton Exchange Membrane (PEM) fuel cells. It is shown through a representative case study that, in theory, fuel cell based power supplies will provide much longer range than the best current rechargeable battery technology. It also briefly discusses an important limitation that prevents fuel cells from achieving their ideal performance, namely a practical method to store their fuel (hydrogen) in a form that is compatible with small mobile field robots. A very efficient Fig. 1. Example of small robots (Left) A ball shaped hopping robot concept fuel storage concept based on water activated lithium hydride for exploration of extreme terrains and caves developed for NASA. (Right) (LiH) is proposed that releases hydrogen on demand. This iRobot 110 Firstlook used for observation, security and search and rescue. concept is very attractive because water vapor from the air is passively extracted or waste water from the fuel cell is recycled and transferred to the lithium hydride where the hydrogen is the near future. Hence, new means for powering field robots “stripped” from water and is returned to the fuel cell to form more water. This results in higher hydrogen storage efficiencies need to be considered. than conventional storage methods. Experimental results are This paper explores the use of fuel cells, and in particular presented that demonstrate the effectiveness of the approach. -

Hydrogen Generation from Magnesium Hydride by Using Organic Acid Yen-Hsi Ho University of Wisconsin-Milwaukee
University of Wisconsin Milwaukee UWM Digital Commons Theses and Dissertations August 2013 Hydrogen Generation from Magnesium Hydride By Using Organic Acid Yen-Hsi Ho University of Wisconsin-Milwaukee Follow this and additional works at: https://dc.uwm.edu/etd Part of the Mechanical Engineering Commons, and the Oil, Gas, and Energy Commons Recommended Citation Ho, Yen-Hsi, "Hydrogen Generation from Magnesium Hydride By Using Organic Acid" (2013). Theses and Dissertations. 286. https://dc.uwm.edu/etd/286 This Thesis is brought to you for free and open access by UWM Digital Commons. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of UWM Digital Commons. For more information, please contact [email protected]. HYDROGEN GENERATION FROM MAGNESIUM HYDRIDE BY USING ORGANIC ACID by Yen-Hsi Ho A Thesis Submitted in Partial Fulfillment of the Requirements for the Degree of Master of Science in Engineering at The University of Wisconsin-Milwaukee August 2013 ABSTRACT HYDROGEN GENERATION FROM MAGNESIUM HYDRIDE BY USING ORGANIC ACID by Yen-Hsi Ho The University of Wisconsin-Milwaukee, 2013 Under the Supervision of Professor Tien-Chien Jen In this paper, the hydrolysis of solid magnesium hydride has been studied with the high concentration of catalyst at the varying temperature. An organic acid (acetic acid, CH3COOH) has been chosen as the catalyst. The study has three objectives: first, using three different weights of MgH2 react with aqueous solution of acid for the hydrogen generation experiments. Secondly, utilizing acetic acid as the catalyst accelerates hydrogen generation. Third, emphasizing the combination of the three operating conditions (the weight of MgH2, the concentration of acetic acid, and the varying temperature) influence the amount of hydrogen generation.