First-Principles-Based Simulations for G Protein-Coupled Receptor Activation and for Large-Scale Nonadiabatic Electron Dynamics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Strategies to Increase ß-Cell Mass Expansion
This electronic thesis or dissertation has been downloaded from the King’s Research Portal at https://kclpure.kcl.ac.uk/portal/ Strategies to increase -cell mass expansion Drynda, Robert Lech Awarding institution: King's College London The copyright of this thesis rests with the author and no quotation from it or information derived from it may be published without proper acknowledgement. END USER LICENCE AGREEMENT Unless another licence is stated on the immediately following page this work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International licence. https://creativecommons.org/licenses/by-nc-nd/4.0/ You are free to copy, distribute and transmit the work Under the following conditions: Attribution: You must attribute the work in the manner specified by the author (but not in any way that suggests that they endorse you or your use of the work). Non Commercial: You may not use this work for commercial purposes. No Derivative Works - You may not alter, transform, or build upon this work. Any of these conditions can be waived if you receive permission from the author. Your fair dealings and other rights are in no way affected by the above. Take down policy If you believe that this document breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 02. Oct. 2021 Strategies to increase β-cell mass expansion A thesis submitted by Robert Drynda For the degree of Doctor of Philosophy from King’s College London Diabetes Research Group Division of Diabetes & Nutritional Sciences Faculty of Life Sciences & Medicine King’s College London 2017 Table of contents Table of contents ................................................................................................. -

Transcriptomic Profiling of Pancreatic Alpha, Beta and Delta Cell Populations Identifies Delta Cells As a Principal Target for Ghrelin in Mouse Islets
Diabetologia (2016) 59:2156–2165 DOI 10.1007/s00125-016-4033-1 ARTICLE Transcriptomic profiling of pancreatic alpha, beta and delta cell populations identifies delta cells as a principal target for ghrelin in mouse islets Alice E. Adriaenssens1 & Berit Svendsen2,3 & Brian Y. H. Lam1 & Giles S. H. Yeo1 & Jens J. Holst2,3 & Frank Reimann1 & Fiona M. Gribble 1 Received: 15 March 2016 /Accepted: 1 June 2016 /Published online: 7 July 2016 # The Author(s) 2016. This article is published with open access at Springerlink.com Abstract using islets with delta cell restricted expression of the calcium Aims/hypothesis Intra-islet and gut–islet crosstalk are critical reporter GCaMP3, and in perfused mouse pancreases. in orchestrating basal and postprandial metabolism. The aim Results A database was constructed of all genes expressed in of this study was to identify regulatory proteins and receptors alpha, beta and delta cells. The gene encoding the ghrelin underlying somatostatin secretion though the use of receptor, Ghsr, was highlighted as being highly expressed transcriptomic comparison of purified murine alpha, beta and enriched in delta cells. Activation of the ghrelin receptor and delta cells. raised cytosolic calcium levels in primary pancreatic delta Methods Sst-Cre mice crossed with fluorescent reporters were cells and enhanced somatostatin secretion in perfused used to identify delta cells, while Glu-Venus (with Venus re- pancreases, correlating with a decrease in insulin and gluca- ported under the control of the Glu [also known as Gcg]pro- gon release. The inhibition of insulin secretion by ghrelin was moter) mice were used to identify alpha and beta cells. -

Guthrie Cdna Resource Center
cDNA Resource Center cDNA Resource Center Catalog cDNA Resource Center Missouri University of Science and Technology 400 W 11th Rolla, MO 65409 TEL: (573) 341-7610 FAX: (573) 341-7609 EMAIL: [email protected] www.cdna.org September, 2008 1 cDNA Resource Center Visit our web site for product updates 2 cDNA Resource Center The cDNA Resource Center The cDNA Resource Center is a service provided by the faculty of the Department of Biological Sciences of Missouri University of Science and Technology. The purpose of the cDNA Resource Center is to further scientific investigation by providing cDNA clones of human proteins involved in signal transduction processes. This is achieved by providing high quality clones for important signaling proteins in a timely manner. By high quality, we mean that the clones are • Sequence verified • Propagated in a versatile vector useful in bacterial and mammalian systems • Free of extraneous 3' and 5' untranslated regions • Expression verified (in most cases) by coupled in vitro transcription/translation assays • Available in wild-type, epitope-tagged and common mutant forms (e.g., constitutively- active or dominant negative) By timely, we mean that the clones are • Usually shipped within a day from when you place your order. Clones can be ordered from our web pages, by FAX or by phone. Within the United States, clones are shipped by overnight courier (FedEx); international orders are shipped International Priority (FedEx). The clones are supplied for research purposes only. Details on use of the material are included on the Material Transfer Agreement (page 3). Clones are distributed by agreement in Invitrogen's pcDNA3.1+ vector. -

Quantigene Flowrna Probe Sets Currently Available
QuantiGene FlowRNA Probe Sets Currently Available Accession No. Species Symbol Gene Name Catalog No. NM_003452 Human ZNF189 zinc finger protein 189 VA1-10009 NM_000057 Human BLM Bloom syndrome VA1-10010 NM_005269 Human GLI glioma-associated oncogene homolog (zinc finger protein) VA1-10011 NM_002614 Human PDZK1 PDZ domain containing 1 VA1-10015 NM_003225 Human TFF1 Trefoil factor 1 (breast cancer, estrogen-inducible sequence expressed in) VA1-10016 NM_002276 Human KRT19 keratin 19 VA1-10022 NM_002659 Human PLAUR plasminogen activator, urokinase receptor VA1-10025 NM_017669 Human ERCC6L excision repair cross-complementing rodent repair deficiency, complementation group 6-like VA1-10029 NM_017699 Human SIDT1 SID1 transmembrane family, member 1 VA1-10032 NM_000077 Human CDKN2A cyclin-dependent kinase inhibitor 2A (melanoma, p16, inhibits CDK4) VA1-10040 NM_003150 Human STAT3 signal transducer and activator of transcripton 3 (acute-phase response factor) VA1-10046 NM_004707 Human ATG12 ATG12 autophagy related 12 homolog (S. cerevisiae) VA1-10047 NM_000737 Human CGB chorionic gonadotropin, beta polypeptide VA1-10048 NM_001017420 Human ESCO2 establishment of cohesion 1 homolog 2 (S. cerevisiae) VA1-10050 NM_197978 Human HEMGN hemogen VA1-10051 NM_001738 Human CA1 Carbonic anhydrase I VA1-10052 NM_000184 Human HBG2 Hemoglobin, gamma G VA1-10053 NM_005330 Human HBE1 Hemoglobin, epsilon 1 VA1-10054 NR_003367 Human PVT1 Pvt1 oncogene homolog (mouse) VA1-10061 NM_000454 Human SOD1 Superoxide dismutase 1, soluble (amyotrophic lateral sclerosis 1 (adult)) -

Biased Signaling of G Protein Coupled Receptors (Gpcrs): Molecular Determinants of GPCR/Transducer Selectivity and Therapeutic Potential
Pharmacology & Therapeutics 200 (2019) 148–178 Contents lists available at ScienceDirect Pharmacology & Therapeutics journal homepage: www.elsevier.com/locate/pharmthera Biased signaling of G protein coupled receptors (GPCRs): Molecular determinants of GPCR/transducer selectivity and therapeutic potential Mohammad Seyedabadi a,b, Mohammad Hossein Ghahremani c, Paul R. Albert d,⁎ a Department of Pharmacology, School of Medicine, Bushehr University of Medical Sciences, Iran b Education Development Center, Bushehr University of Medical Sciences, Iran c Department of Toxicology–Pharmacology, School of Pharmacy, Tehran University of Medical Sciences, Iran d Ottawa Hospital Research Institute, Neuroscience, University of Ottawa, Canada article info abstract Available online 8 May 2019 G protein coupled receptors (GPCRs) convey signals across membranes via interaction with G proteins. Origi- nally, an individual GPCR was thought to signal through one G protein family, comprising cognate G proteins Keywords: that mediate canonical receptor signaling. However, several deviations from canonical signaling pathways for GPCR GPCRs have been described. It is now clear that GPCRs can engage with multiple G proteins and the line between Gprotein cognate and non-cognate signaling is increasingly blurred. Furthermore, GPCRs couple to non-G protein trans- β-arrestin ducers, including β-arrestins or other scaffold proteins, to initiate additional signaling cascades. Selectivity Biased Signaling Receptor/transducer selectivity is dictated by agonist-induced receptor conformations as well as by collateral fac- Therapeutic Potential tors. In particular, ligands stabilize distinct receptor conformations to preferentially activate certain pathways, designated ‘biased signaling’. In this regard, receptor sequence alignment and mutagenesis have helped to iden- tify key receptor domains for receptor/transducer specificity. -

The Dominant Somatostatin Receptor in Neuroendocrine Tumors of North Indian Population 1Narendra Krishnani, 2Niraj Kumari, 3Rajneesh K Singh, 4Pooja Shukla
WJOES Narendra Krishnani et al 10.5005/jp-journals-10002-1171 ORIGINAL ARTICLE The Dominant Somatostatin Receptor in Neuroendocrine Tumors of North Indian Population 1Narendra Krishnani, 2Niraj Kumari, 3Rajneesh K Singh, 4Pooja Shukla ABSTRACT Neuroendocrine Tumors of North Indian Population. World J Endoc Surg 2015;7(3):60-64. Introduction: Neuroendocrine tumors (NET) express diffe rent types of somatostatin receptors (SSTRs) that bind to syn Source of support: Nil thetic analogs with variable affinity. It is important to know the Conflict of interest: None expression profile of SSTRs to predict biological effect of somato- statin analogues. We studied SSTR2 and SSTR5 expre ssion by immunohistochemistry (IHC) to assess the dominant sub INTRODUCTION type in NETs and correlate the expression with histological Neuroendocrine tumors (NET) are heterogeneous group prognostic parameters. of neoplasms that arise primarily in gastrointestinal tract Materials and methods: Fiftythree consecutive cases of NET (GIT), pancreas and lung.1 Ninety percent of these tumors from all sites were evaluated for SSTR2 and SSTR5 expres are nonfunctional, that is, they do not produce bio- sion by IHC. The expression was correlated with histological features of NETs. logically active peptides but are diagnosed late because of their mass effect.2 A common feature of all NETs is Results: Fortyfour cases were resected specimens and 9 were small biopsies. Nine of 53 cases (16.9%) were functional expression of different types of somatostatin receptors tumors. There were 24 NETs from gastrointestinal tract (GIT), (SSTRs) which are seen in approximately 80 to 90% of 19 from pancreas and 10 from miscellaneous sites. -

General Summary I Alvarado, F Sissingh and J Licinio
Molecular Psychiatry (2002) 7, 665–668 2002 Nature Publishing Group All rights reserved 1359-4184/02 $25.00 www.nature.com/mp General Summary I Alvarado, F Sissingh and J Licinio UCLA, Los Angeles, CA, USA Molecular Psychiatry (2002) 7, 665–668. doi:10.1038/ teins of epidermal growth factor (EGF) and its homol- sj.mp.4001225 ogues are known to regulate development of the dopa- minergic neurons as well as dopamine metabolism, and might be involved in schizophrenic pathology and/or etiology. To test this possibility, levels of these SCIENTIFIC CORRESPONDENCE proteins and their receptors were determined in post- Association of a polymorphism in the promoter mortem brains of schizophrenic patients and control region of the serotonin 5-HT2C receptor gene with subjects in the present study. Among members of the tardive dyskinesia in patients with schizophrenia EGF family examined, EGF were specifically decreased ZJ Zhang, XB Zhang, WW Sha, XB Zhang, in the prefrontal cortex and striatum, where the dopa- GP Reynolds minergic neurons innervate. Conversely, receptor lev- els for EGF were increased in the same region. More- Tardive dyskinesia (TD) is a major side effect of over, the reduction in EGF was also seen in serum of chronic treatment with antipsychotic drugs. As the 5- schizophrenic patients, even in drug-naive patients. HT2C receptor is involved in motor function including However, chronic administration of haloperidol to rats production of dyskinesias, the authors have investi- had no effect on EGF levels. These findings suggest a gated whether functional polymorphisms of the pro- strong link between abnormal EGF receptor signaling moter region of the 5-HT2C receptor gene may be asso- and schizophrenia that might underlie the domami- ciated with TD. -
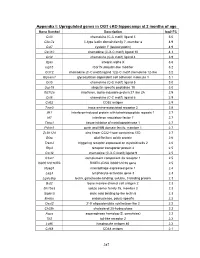
PDF (Appendices)
Appendix I: Upregulated genes in OGT cKO hippocampi at 2 months of age Gene Symbol Description log2 FC Ccl3 chemokine (C-C motif) ligand 3 5.0 Clec7a C-type lectin domain family 7, member a 4.9 Cst7 cystatin F (leukocystatin) 4.9 Cxcl10 chemokine (C-X-C motif) ligand 10 4.3 Ccl4 chemokine (C-C motif) ligand 4 3.9 Itgax integrin alpha X 3.6 Isg15 ISG15 ubiquitin-like modifier 3.2 Ccl12 chemokine (C-C motif) ligand 12|c-C motif chemokine 12-like 3.2 Glycam1 glycosylation dependent cell adhesion molecule 1 3.1 Ccl5 chemokine (C-C motif) ligand 5 3.0 Usp18 ubiquitin specific peptidase 18 3.0 Ifi27l2a interferon, alpha-inducible protein 27 like 2A 2.9 Ccl6 chemokine (C-C motif) ligand 6 2.9 Cd52 CD52 antigen 2.9 Taar3 trace amine-associated receptor 3 2.8 Ifit1 interferon-induced protein with tetratricopeptide repeats 1 2.7 Irf7 interferon regulatory factor 7 2.7 Timp1 tissue inhibitor of metalloproteinase 1 2.7 Pyhin1 pyrin and HIN domain family, member 1 2.7 Zc3h12d zinc finger CCCH type containing 12D 2.7 Gfap glial fibrillary acidic protein 2.6 Trem2 triggering receptor expressed on myeloid cells 2 2.6 Rtp4 receptor transporter protein 4 2.5 Cxcl9 chemokine (C-X-C motif) ligand 9 2.5 C3ar1 complement component 3a receptor 1 2.5 I830012O16Rik RIKEN cDNA I830012O16 gene 2.5 Mpeg1 macrophage expressed gene 1 2.4 Lag3 lymphocyte-activation gene 3 2.3 Lgals3bp lectin, galactoside-binding, soluble, 3 binding protein 2.3 Bst2 bone marrow stromal cell antigen 2 2.3 Slc15a3 solute carrier family 15, member 3 2.3 Siglec5 sialic acid binding Ig-like lectin 5 2.3 Endou endonuclease, polyU-specific 2.2 Oasl2 2'-5' oligoadenylate synthetase-like 2 2.2 Ch25h cholesterol 25-hydroxylase 2.2 Aspg asparaginase homolog (S. -

Establishing Stable Cell Lines
Establishing Stable Cell Lines 1. Our proprietary expression system for mammalian cells is used. 2. Our stable cell lines are from the single colonies, not from the pool. 3. Many single clones are screened, and three best ones are kept. 4. Functional assay is performed to confirm the clones’ biological function. 5. Time frame: 2 – 3 months for one stable cell line List of In-Stock ACTOne GPCR Stable Clones Transduced Gi-coupled receptors (22) Transduced Gs coupled receptors (34) Cannabinoid receptor 1 (CB1) Vasoactive Intestinal Peptide Receptor 2 (VIPR2) Dopamine Receptor 2 (DRD2) Melanocortin 4 Receptor (MC4R) Melanocortin 5 Receptor (MC5R) Somatostatin Receptor 5 (SSTR5) Parathyroid Hormone Receptor 1 (PTHR1) Adenosine A1 Receptor (ADORA1) Glucagon Receptor (GCGR) Chemokine (C-C motif) receptor 5 (CCR5) Dopamine Receptor 1 (DRD1) Melanin-concentrating Hormone Receptor 1 (MCHR1) Prostaglandin E Receptor 4 (EP4) Vasoactive Intestinal Peptide Receptor 1 (VIPR1) Cannabinoid receptor 2 (CB2) Gastric Inhibitor Peptide Receptor (GIPR) Glutamate receptor, metabotropic 8 (GRM8) Dopamine Receptor 5 (DRD5) Opioid receptor, kappa 1 (OPRK1) Parathyroid Hormone Receptor 2 (PTHR2) Adenosine A3 receptor (ADORA3) 5-hydroxytryptamine (serotonin) receptor 6 (HTR4) Corticotropin Releasing Hormone Receptor 2 (CRHR2) Glutamate receptor, metabotropic 8 (GRM8) Adenylate Cyclase Activating Polypeptide 1 Receptor type I (ADCYAP1R1) Neuropeptide Y Receptor Y1 (NPY1R) Secretin Receptor (SCTR) Neuropeptide Y Receptor Y2 (NPY2R) Follicle -

I Chemical Biology Approaches to Combat Parkinson's Disease By
Chemical Biology Approaches to Combat Parkinson’s Disease by Felix O. Nwogbo Jr Department of Chemistry Duke University Date: _______________________ Approved: ___________________________ Dewey G. McCafferty, Supervisor ___________________________ Jennifer L. Roizen ___________________________ Jiyong Hong ___________________________ David M. Gooden Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Chemistry in the Graduate School of Duke University 2018 i v ABSTRACT Chemical Biology Approaches to Combat Parkinson’s Disease by Felix O. Nwogbo Jr Department of Chemistry Duke University Date: _______________________ Approved: ___________________________ Dewey G. McCafferty, Supervisor ___________________________ Jennifer L. Roizen ___________________________ Jiyong Hong ___________________________ David M. Gooden An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Chemistry in the Graduate School of Duke University 2018 i v Copyright by Felix O. Nwogbo Jr 2018 Abstract Parkinson's disease (PD) is a debilitating neurodegenerative disease of the central nervous system characterized by loss of striatal dopaminergic projections from the substantia nigra. Although there is no cure for PD, dopamine (DA) replacement using L-3,4-dihydroxyphenylalanine (L-DOPA) is the most common therapy used to manage PD motor symptoms. L-DOPA is poorly absorbed into the brain and metabolized in the -

Dynamic Mass Redistribution Reveals Diverging Importance of PDZ
Pharmacological Research 105 (2016) 13–21 Contents lists available at ScienceDirect Pharmacological Research j ournal homepage: www.elsevier.com/locate/yphrs Dynamic mass redistribution reveals diverging importance of PDZ-ligands for G protein-coupled receptor pharmacodynamics a b b b Nathan D. Camp , Kyung-Soon Lee , Allison Cherry , Jennifer L. Wacker-Mhyre , b b b b Timothy S. Kountz , Ji-Min Park , Dorathy-Ann Harris , Marianne Estrada , b b a b,∗ Aaron Stewart , Nephi Stella , Alejandro Wolf-Yadlin , Chris Hague a Department of Genome Sciences, University of Washington School of Medicine, Seattle, WA 98195, USA b Department of Pharmacology, University of Washington School of Medicine, Seattle, WA 98195, USA a r t i c l e i n f o a b s t r a c t Article history: G protein-coupled receptors (GPCRs) are essential membrane proteins that facilitate cell-to-cell Received 19 October 2015 communication and co-ordinate physiological processes. At least 30 human GPCRs contain a Type I PSD- Received in revised form 95/DLG/Zo-1 (PDZ) ligand in their distal C-terminal domain; this four amino acid motif of X-[S/T]-X-[] 28 December 2015 sequence facilitates interactions with PDZ domain-containing proteins. Because PDZ protein interactions Accepted 1 January 2016 have profound effects on GPCR ligand pharmacology, cellular localization, signal-transduction effector Available online 7 January 2016 coupling and duration of activity, we analyzed the importance of Type I PDZ ligands for the function of 23 full-length and PDZ-ligand truncated (PDZ) human GPCRs in cultured human cells. SNAP-epitope tag Keywords: polyacrylamide gel electrophoresis revealed most Type I PDZ GPCRs exist as both monomers and mul- G protein-coupled receptor timers; removal of the PDZ ligand played minimal role in multimer formation. -

SUPPLEMENTARY APPENDIX Exome Sequencing Reveals Heterogeneous Clonal Dynamics in Donor Cell Myeloid Neoplasms After Stem Cell Transplantation
SUPPLEMENTARY APPENDIX Exome sequencing reveals heterogeneous clonal dynamics in donor cell myeloid neoplasms after stem cell transplantation Julia Suárez-González, 1,2 Juan Carlos Triviño, 3 Guiomar Bautista, 4 José Antonio García-Marco, 4 Ángela Figuera, 5 Antonio Balas, 6 José Luis Vicario, 6 Francisco José Ortuño, 7 Raúl Teruel, 7 José María Álamo, 8 Diego Carbonell, 2,9 Cristina Andrés-Zayas, 1,2 Nieves Dorado, 2,9 Gabriela Rodríguez-Macías, 9 Mi Kwon, 2,9 José Luis Díez-Martín, 2,9,10 Carolina Martínez-Laperche 2,9* and Ismael Buño 1,2,9,11* on behalf of the Spanish Group for Hematopoietic Transplantation (GETH) 1Genomics Unit, Gregorio Marañón General University Hospital, Gregorio Marañón Health Research Institute (IiSGM), Madrid; 2Gregorio Marañón Health Research Institute (IiSGM), Madrid; 3Sistemas Genómicos, Valencia; 4Department of Hematology, Puerta de Hierro General University Hospital, Madrid; 5Department of Hematology, La Princesa University Hospital, Madrid; 6Department of Histocompatibility, Madrid Blood Centre, Madrid; 7Department of Hematology and Medical Oncology Unit, IMIB-Arrixaca, Morales Meseguer General University Hospital, Murcia; 8Centro Inmunológico de Alicante - CIALAB, Alicante; 9Department of Hematology, Gregorio Marañón General University Hospital, Madrid; 10 Department of Medicine, School of Medicine, Com - plutense University of Madrid, Madrid and 11 Department of Cell Biology, School of Medicine, Complutense University of Madrid, Madrid, Spain *CM-L and IB contributed equally as co-senior authors. Correspondence: