Characterisation of L-Cell Secretory Mechanisms and Colonic
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Strategies to Increase ß-Cell Mass Expansion
This electronic thesis or dissertation has been downloaded from the King’s Research Portal at https://kclpure.kcl.ac.uk/portal/ Strategies to increase -cell mass expansion Drynda, Robert Lech Awarding institution: King's College London The copyright of this thesis rests with the author and no quotation from it or information derived from it may be published without proper acknowledgement. END USER LICENCE AGREEMENT Unless another licence is stated on the immediately following page this work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International licence. https://creativecommons.org/licenses/by-nc-nd/4.0/ You are free to copy, distribute and transmit the work Under the following conditions: Attribution: You must attribute the work in the manner specified by the author (but not in any way that suggests that they endorse you or your use of the work). Non Commercial: You may not use this work for commercial purposes. No Derivative Works - You may not alter, transform, or build upon this work. Any of these conditions can be waived if you receive permission from the author. Your fair dealings and other rights are in no way affected by the above. Take down policy If you believe that this document breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 02. Oct. 2021 Strategies to increase β-cell mass expansion A thesis submitted by Robert Drynda For the degree of Doctor of Philosophy from King’s College London Diabetes Research Group Division of Diabetes & Nutritional Sciences Faculty of Life Sciences & Medicine King’s College London 2017 Table of contents Table of contents ................................................................................................. -

The Activation of the Glucagon-Like Peptide-1 (GLP-1) Receptor by Peptide and Non-Peptide Ligands
The Activation of the Glucagon-Like Peptide-1 (GLP-1) Receptor by Peptide and Non-Peptide Ligands Clare Louise Wishart Submitted in accordance with the requirements for the degree of Doctor of Philosophy of Science University of Leeds School of Biomedical Sciences Faculty of Biological Sciences September 2013 I Intellectual Property and Publication Statements The candidate confirms that the work submitted is her own and that appropriate credit has been given where reference has been made to the work of others. This copy has been supplied on the understanding that it is copyright material and that no quotation from the thesis may be published without proper acknowledgement. The right of Clare Louise Wishart to be identified as Author of this work has been asserted by her in accordance with the Copyright, Designs and Patents Act 1988. © 2013 The University of Leeds and Clare Louise Wishart. II Acknowledgments Firstly I would like to offer my sincerest thanks and gratitude to my supervisor, Dr. Dan Donnelly, who has been nothing but encouraging and engaging from day one. I have thoroughly enjoyed every moment of working alongside him and learning from his guidance and wisdom. My thanks go to my academic assessor Professor Paul Milner whom I have known for several years, and during my time at the University of Leeds he has offered me invaluable advice and inspiration. Additionally I would like to thank my academic project advisor Dr. Michael Harrison for his friendship, help and advice. I would like to thank Dr. Rosalind Mann and Dr. Elsayed Nasr for welcoming me into the lab as a new PhD student and sharing their experimental techniques with me, these techniques have helped me no end in my time as a research student. -

1488.Full.Pdf
Downloaded from genesdev.cshlp.org on September 26, 2021 - Published by Cold Spring Harbor Laboratory Press Neurogenin 3 is essential for the proper specification of gastric enteroendocrine cells and the maintenance of gastric epithelial cell identity Catherine S. Lee,1 Nathalie Perreault,1 John E. Brestelli, and Klaus H. Kaestner2 Department of Genetics, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania 19104, USA The notch signaling pathway is essential for the endocrine cell fate in various tissues including the enteroendocrine system of the gastrointestinal tract. Enteroendocrine cells are one of the four major cell types found in the gastric epithelium of the glandular stomach. To understand the molecular basis of enteroendocrine cell development, we have used gene targeting in mouse embryonic stem cells to derive an EGFP-marked null allele of the bHLH transcription factor, neurogenin 3 (ngn3). In ngn3−/− mice, glucagon secreting A-cells, somatostatin secreting D-cells, and gastrin secreting G-cells are absent from the epithelium of the glandular stomach, whereas the number of serotonin-expressing enterochromaffin (EC) cells is decreased dramatically. In addition, ngn3−/− mice display intestinal metaplasia of the gastric epithelium. Thus, ngn3 is required for the differentiation of enteroendocrine cells in the stomach and the maintenance of gastric epithelial cell identity. [Key Words: Basic-helix-loop-helix (bHLH) protein; neurogenin 3 (ngn3); notch signaling; metaplasia; enteroendocrine cells; iFABP; Muc 2] Received February 15, 2002; revised version accepted May 2, 2002. The mouse stomach is divided into two domains, the tors including the neurogenin genes (Sommer et al. 1996; proximal third, which is known as the forestomach and Artavanis-Tsakonas et al. -
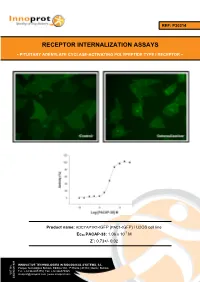
Receptor Internalization Assays
REF: P30214 RECEPTOR INTERNALIZATION ASSAYS - PITUITARY ADENYLATE CYCLASE-ACTIVATING POLYPEPTIDE TYPE I RECEPTOR - Product name: ADCYAP1R1-tGFP (PAC1-tGFP) / U2OS cell line -7 Ec50 PACAP-38: 1.06 x 10 M Z´: 0.73+/- 0.02 INNOVATIVE TECHNOLOGIES IN BIOLOGICAL SYSTEMS, S.L. Parque Tecnológico Bizkaia, Edifício 502, 1ª Planta | 48160 | Derio | Bizkaia Tel.: +34 944005355 | Fax: +34 946579925 VAT No. [email protected] | www.innoprot.com ESB95481909 Product Name: ADCYAP1R1-tGFP_U2OS Reference: P30214 Rep. Official Full Name: Pituitary adenylate cyclase- activating polypeptide type I receptor DNA Accession Number: Gene Bank AY366498 Host Cell: U2OS References: P30214: 2 vials of 3 x 106 proliferative cells P30214-DA: 1 vial of 2 x 106 division-arrested cells Storage: Liquid Nitrogen Assay Briefly description About ADCYAP1R1 Each vial of ADCYAP1R1 Internalization Assay Pituitary adenylate cyclase-activating Cell Line contains U2OS cells stably expressing polypeptide type I receptor, also known as human Pituitary adenylate cyclase-activating PAC1 is a protein that in humans is encoded by polypeptide type I receptor tagged in the N- the ADCYAP1R1 gene. ADCYAP1R1 is a terminus with tGFP protein. membrane-associated protein and shares significant homology with members of the Innoprot’s ADCYAP1R1-tGFP Internalization glucagon/secretin receptor family. This receptor Assay Cell Line has been designed to assay binds pituitary adenylate cyclase activating potential agonists/ antagonists against peptide (PACAP) mediating several biological ADCYAP1R1, modulating its activation and the activities and it is positively coupled to following redistribution process inside the cells. adenylate cyclase. This cell line will allow the image analysis of the stimuli induced by the compounds. -

Multiplex Gpcr Internalization Assay Using Reverse Transduction on Adenoviral Vector Immobilized Microparticles S
MULTIPLEX GPCR INTERNALIZATION ASSAY USING REVERSE TRANSDUCTION ON ADENOVIRAL VECTOR IMMOBILIZED MICROPARTICLES S. Han1,2, H.J. Bae1,2, W. Park3 and S. Kwon1,2* 1Department of Electrical and Computer Engineering, Inter-university Semiconductor Research Center (ISRC), Seoul National University, SOUTH KOREA 2Center for Nanoparticle Research, Institute for Basic Science (IBS), SOUTH KOREA and 3Department of Electronics and Radio Engineering, Institute for Laser Engineering, Kyung Hee University, SOUTH KOREA ABSTRACT We present a new multiplexing method for high-throughput cell-based assays in a microtiter well based on reverse transduction of cells by adenoviral vectors immobilized on encoded microparticles. Our particle-based approach spatially confines the gene delivery to cells seeded on the particles and provides the code for identifying the delivered gene, thus easily achieving a multiplex cell microarray in a microtiter well by means of a single pipetting without the cross- expression of genes and the positional identification. Utilizing this method with adenoviral vectors having a G-protein coupled recpeptor (GPCR) gene, we demonstrated 3-plex GPCR internalization assay. KEYWORDS: Multiplex GPCR assay, Reverse transduction, Adenovirus, Encoded microparticle INTRODUCTION G-protein coupled receptors (GPCRs) in the cell membrane are major drug targets in pharmaceutical industry since they interact with a huge variety of endogenous ligands and trigger intracellular functions related to many physiological processes or diseases [1]. Many cell-based assay strategies have been developed to identify GPCR-targeted drugs with more biologically relevant data. Since typical cell-based assays are performed in the microtiter wells and it allows only one type of receptor for each well, multiplex cellular assay technologies have emerged to run high-throughput compound screening with over several hundreds of GPCRs. -

Autism and Gastrointestinal Symptoms Karoly Horvath, MD, Phd and Jay A
Autism and Gastrointestinal Symptoms Karoly Horvath, MD, PhD and Jay A. Perman, MD Address In the last decade, the focus in autism research migrated Department of Pediatrics, University of Maryland School of Medicine, from psychological studies to exploration of the biologic 22 South Greene Street, N5W70, Box 140, Baltimore, basis of this devastating disorder. Studies using neuro- MD 21201-1595, USA. E-mail: [email protected] imaging and brain autopsy, as well as immunologic, genetic, metabolic, and gastrointestinal research efforts, have Current Gastroenterology Reports 2002, 4:251–258 Current Science Inc. ISSN 1522–8037 resulted in a significant amount of new information. Copyright © 2002 by Current Science Inc. However, this biologic research is still in the evolutionary stage, with many controversies, especially in brain and genetic research. For example, no consensus has been Autism is a collection of behavioral symptoms reached regarding the brain areas responsible for autism. characterized by dysfunction in social interaction and The gastrointestinal tract is an easier target for investi- communication in affected children. It is typically gation than the brain. However, only two studies of associated with restrictive, repetitive, and stereotypic gastrointestinal symptoms in autism were reported prior behavior and manifests within the first 3 years of life. to 1996. In 1971, a report of 15 randomly selected autistic The cause of this disorder is not known. Over the patients described six children who had bulky, odorous, past decade, a significant upswing in research has or loose stools, or intermittent diarrhea, and one with celiac occurred to examine the biologic basis of autism. disease [2]. -

Molecular Dissection of G-Protein Coupled Receptor Signaling and Oligomerization
MOLECULAR DISSECTION OF G-PROTEIN COUPLED RECEPTOR SIGNALING AND OLIGOMERIZATION BY MICHAEL RIZZO A Dissertation Submitted to the Graduate Faculty of WAKE FOREST UNIVERSITY GRADUATE SCHOOL OF ARTS AND SCIENCES in Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY Biology December, 2019 Winston-Salem, North Carolina Approved By: Erik C. Johnson, Ph.D. Advisor Wayne E. Pratt, Ph.D. Chair Pat C. Lord, Ph.D. Gloria K. Muday, Ph.D. Ke Zhang, Ph.D. ACKNOWLEDGEMENTS I would first like to thank my advisor, Dr. Erik Johnson, for his support, expertise, and leadership during my time in his lab. Without him, the work herein would not be possible. I would also like to thank the members of my committee, Dr. Gloria Muday, Dr. Ke Zhang, Dr. Wayne Pratt, and Dr. Pat Lord, for their guidance and advice that helped improve the quality of the research presented here. I would also like to thank members of the Johnson lab, both past and present, for being valuable colleagues and friends. I would especially like to thank Dr. Jason Braco, Dr. Jon Fisher, Dr. Jake Saunders, and Becky Perry, all of whom spent a great deal of time offering me advice, proofreading grants and manuscripts, and overall supporting me through the ups and downs of the research process. Finally, I would like to thank my family, both for instilling in me a passion for knowledge and education, and for their continued support. In particular, I would like to thank my wife Emerald – I am forever indebted to you for your support throughout this process, and I will never forget the sacrifices you made to help me get to where I am today. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Vocabulario De Morfoloxía, Anatomía E Citoloxía Veterinaria
Vocabulario de Morfoloxía, anatomía e citoloxía veterinaria (galego-español-inglés) Servizo de Normalización Lingüística Universidade de Santiago de Compostela COLECCIÓN VOCABULARIOS TEMÁTICOS N.º 4 SERVIZO DE NORMALIZACIÓN LINGÜÍSTICA Vocabulario de Morfoloxía, anatomía e citoloxía veterinaria (galego-español-inglés) 2008 UNIVERSIDADE DE SANTIAGO DE COMPOSTELA VOCABULARIO de morfoloxía, anatomía e citoloxía veterinaria : (galego-español- inglés) / coordinador Xusto A. Rodríguez Río, Servizo de Normalización Lingüística ; autores Matilde Lombardero Fernández ... [et al.]. – Santiago de Compostela : Universidade de Santiago de Compostela, Servizo de Publicacións e Intercambio Científico, 2008. – 369 p. ; 21 cm. – (Vocabularios temáticos ; 4). - D.L. C 2458-2008. – ISBN 978-84-9887-018-3 1.Medicina �������������������������������������������������������������������������veterinaria-Diccionarios�������������������������������������������������. 2.Galego (Lingua)-Glosarios, vocabularios, etc. políglotas. I.Lombardero Fernández, Matilde. II.Rodríguez Rio, Xusto A. coord. III. Universidade de Santiago de Compostela. Servizo de Normalización Lingüística, coord. IV.Universidade de Santiago de Compostela. Servizo de Publicacións e Intercambio Científico, ed. V.Serie. 591.4(038)=699=60=20 Coordinador Xusto A. Rodríguez Río (Área de Terminoloxía. Servizo de Normalización Lingüística. Universidade de Santiago de Compostela) Autoras/res Matilde Lombardero Fernández (doutora en Veterinaria e profesora do Departamento de Anatomía e Produción Animal. -

EGFR Confers Exquisite Specificity of Wnt9a-Fzd9b Signaling in Hematopoietic Stem Cell Development
bioRxiv preprint doi: https://doi.org/10.1101/387043; this version posted August 7, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Grainger, et al, 2018 EGFR confers exquisite specificity of Wnt9a-Fzd9b signaling in hematopoietic stem cell development Stephanie Grainger1, Nicole Nguyen1, Jenna Richter1,2, Jordan Setayesh1, Brianna Lonquich1, Chet Huan Oon1, Jacob M. Wozniak2,3,4, Rocio Barahona1, Caramai N. Kamei5, Jack Houston1,2, Marvic Carrillo-Terrazas3,4, Iain A. Drummond5,6, David Gonzalez3.4, Karl Willert#,¥,1, and David Traver¥,1,7. ¥co-corresponding authors: [email protected]; [email protected] #Lead contact 1Department of Cellular and Molecular Medicine, University of California, San Diego, La Jolla, California, 92037, USA. 2Biomedical Sciences Graduate Program, University of California, San Diego, La Jolla, California, 92037, USA. 3Skaggs School of Pharmacy and Pharmaceutical Science, University of California, San Diego, La Jolla, California, 92093, USA. 4Department of Pharmacology, University of California, San Diego, La Jolla, California, 92092 5Massachusetts General Hospital Nephrology Division, Charlestown, Massachusetts, 02129, USA. 6Harvard Medical School, Department of Genetics, Boston MA 02115 7Section of Cell and Developmental Biology, University of California, San Diego, La Jolla, California, 92037, USA. Running title: A mechanism for Wnt-Fzd specificity in hematopoietic stem cells Keywords: hematopoietic stem cell (HSC), Wnt, Wnt9a, human, zebrafish, Fzd, Fzd9b, FZD9, EGFR, APEX2 1 bioRxiv preprint doi: https://doi.org/10.1101/387043; this version posted August 7, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

A2B Adenosine Receptors and T Cell Activation 493
Journal of Cell Science 112, 491-502 (1999) 491 Printed in Great Britain © The Company of Biologists Limited 1999 JCS0069 Expression of A2B adenosine receptors in human lymphocytes: their role in T cell activation Maribel Mirabet1, Carolina Herrera1, Oscar J. Cordero2, Josefa Mallol1, Carmen Lluis1 and Rafael Franco1,* 1Department of Biochemistry and Molecular Biology, Faculty of Chemistry, University of Barcelona, Barcelona, Catalonia, Spain 2Department of Biochemistry and Molecular Biology, Faculty of Biology, University of Santiago de Compostela, Spain *Author for correspondence (e-mail: [email protected]; homepage: www.bq.ub.es/recep/franco.html) Accepted 9 December 1998; published on WWW 25 January 1999 SUMMARY Extracellular adenosine has a key role in the development A2BRs but not of A2A receptors in these human cells. The and function of the cells of the immune system. Many of percentage of A2BR-expressing cells was similar in the the adenosine actions seem to be mediated by specific CD4+ or CD8+ T cell subpopulations. Interestingly surface receptors positively coupled to adenylate cyclase: activation signals delivered by either phytohemagglutinin A2A and A2B. Despite the fact that A2A receptors (A2ARs) or anti-T cell receptor/CD3 complex antibodies led to a can be easily studied due to the availability of the specific significant increase in both the percentage of cells agonist CGS21680, a pharmacological and physiological expressing the receptor and the intensity of the labeling. characterization of adenosine A2B receptors (A2BRs) in These receptors are functional since interleukin-2 lymphocytes has not been possible due to the lack of production in these cells is reduced by NECA but not by R- suitable reagents. -

Molecular Characterization of Clonal Human Renal Forming Cells Cohen-Zontag Osnat , Gershon Rotem , Harari-Steinberg Orit , Kant
bioRxiv preprint doi: https://doi.org/10.1101/2020.03.05.978254; this version posted March 6, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Molecular characterization of clonal human renal forming cells Cohen-Zontag Osnat1,7,*, Gershon Rotem1,7,*, Harari-Steinberg Orit1,7,*, Kanter Itamar4, Omer Dorit1,7 , Pleniceanu Oren1,7, Tam Gal4, Oriel Sarit4, Ben-Hur Herzl8,9 , Katz Guy1,3,5,7, Zohar Dotan2,7, Kalisky Tomer4,#, Dekel Benjamin1, 6,7,#,^, Pode- Shakked Naomi1,3,7,#. 1Pediatric Stem Cell Research Institute, Edmond and Lily Safra Children's Hospital, Sheba Medical Center, Tel-Hashomer, Israel 2Dept of Urology, Sheba Medical Center, Tel-Hashomer, Israel 3The Talpiot Medical Leadership Program, Sheba Medical Center, Tel-Hashomer, Israel 4Faculty of Engineering and Bar-Ilan Institute of Nanotechnology and Advanced Materials (BINA), Bar-Ilan University, Ramat Gan, Israel. 5The Joseph Buchman Gynecology and Maternity Center, Sheba Medical Center, Tel- Hashomer, Israel 6Division of Pediatric Nephrology, Edmond and Lily Safra Children's Hospital, Sheba Medical Center, Tel-Hashomer, Israel 7Sackler Faculty of Medicine, Tel-Aviv University, Tel-Aviv, Israel 8L.E.M. Laboratory of Early Detection, Nes Ziona, Israel 9Department of Obstetrics and Gynecology, Assaf Harofeh Medical Center, Tzrifin, Israel *These first authors contributed equally to this work #These senior authors contributed equally to this work ^Correspondence: Benjamin Dekel MD, PhD Pediatric Stem Cell Research Institute Edmond & Lily Safra Children's Hospital, Sheba Medical Center E-mails: [email protected] or [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2020.03.05.978254; this version posted March 6, 2020.