Gene Expression Profiles Associated with Pediatric Relapsed AML
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

C/Ebpα Is an Essential Collaborator in Hoxa9/Meis1-Mediated Leukemogenesis
C/EBPα is an essential collaborator in Hoxa9/Meis1-mediated leukemogenesis Cailin Collinsa, Jingya Wanga, Hongzhi Miaoa, Joel Bronsteina, Humaira Nawera, Tao Xua, Maria Figueroaa, Andrew G. Munteana, and Jay L. Hessa,b,1 aDepartment of Pathology, University of Michigan, Ann Arbor, MI 48109; and bIndiana University School of Medicine, Indianapolis, IN 46202 Edited* by Louis M. Staudt, National Institutes of Health, Bethesda, MD, and approved May 19, 2014 (received for review February 12, 2014) Homeobox A9 (HOXA9) is a homeodomain-containing transcrip- with Hoxa9. In addition, C/EBP recognition motifs are enriched tion factor that plays a key role in hematopoietic stem cell expan- at Hoxa9 binding sites. sion and is commonly deregulated in human acute leukemias. A C/EBPα is a basic leucine-zipper transcription factor that plays variety of upstream genetic alterations in acute myeloid leukemia a critical role in lineage commitment during hematopoietic dif- −/− (AML) lead to overexpression of HOXA9, almost always in associ- ferentiation (18). Whereas Cebpa mice show complete loss of ation with overexpression of its cofactor meis homeobox 1 (MEIS1). the granulocytic compartment, recent work shows that loss of α A wide range of data suggests that HOXA9 and MEIS1 play a syn- C/EBP in adult HSCs leads to both an increase in the number ergistic causative role in AML, although the molecular mechanisms of functional HSCs and an increase in their proliferative and leading to transformation by HOXA9 and MEIS1 remain elusive. In repopulating capacity (19, 20). Conversely, CEBPA overexpression can promote transdifferentiation of a variety of fibroblastic cells to this study, we identify CCAAT/enhancer binding protein alpha (C/ the myeloid lineage and can induce monocytic differentiation in EBPα) as a critical collaborator required for Hoxa9/Meis1-mediated α MLL-fusion protein-mediated leukemias (21, 22). -

Test Summary Flyer-NGS Panels.Pub
Cancer‐related Mutaon Analysis Next Generaon Gene Sequencing for Myeloid Tesng Assay Summary IU Health Molecular Pathology Laboratory now offers high throughput sequencing for hot spot mutations found in clinically relevant cancer genes. In addition to a general panel of 54 genes, selected panels have been developed for a more tai- lored application in specific cancers. Comparing to single gene assay, these panels offer a more comprehensive and eco- nomic way to assess prognosis and/or treatment options for cancer patients at the initial diagnosis or at the relapse. Orderable Name: Use IU Health Molecular Pathology requisition; Call 317.491.6417 for requisition. Panels include: AML Mutations by NGS ASXL1, CEBPA, DNMT3A, ETV6/TEL, FLT3, HRAS, IDH1, IDH2, KIT, KRAS, MLL, NPM1, NRAS, PHF6, RUNX1, TET2, TP53, WT1 MDS Mutations by NGS ASXL1, ATRX, BCOR, BCORL1, ETV6/TEL, DNMT3A, EZH2, GNAS, IDH1, IDH2, RUNX1, SF3B1, SRSF2, TET2, TP53, U2AF1, ZRSR2 CML Mutations by NGS ABL1 MPN Mutations by NGS ASXL1, BRAF, CALR, CSF3R, EZH2, IKZF1, JAK2, JAK3, KDM6A, KIT, MPL, PDGRA, SETBP1, TET2 CMML Mutations by NGS ASXL1, CBL, CBLB, CBLC, EZH2, RUNX1, TET2, TP53, SRSF2 JMML Mutations by NGS CBL, CBLB, CBLC, HRAS, KRAS, NRAS, PTPN11 ALL Mutations by NGS ABL1, CSF3R, FBXW7, IKZF1, JAK3, KDM6A, NOTCH1 CLL Mutations by NGS MYD88, NOTCH1, SF3B1, TP53 Lymphoma/Myeloma Mutations by NGS BRAF, CDKN2A, CSF3R, FBXW7, HRAS, KRAS, MYD88, NOTCH1, NRAS, SF3B1,TP53 Hematopoietic Neoplasms Mutations by NGS ABL1, ASXL1, ATRX, BCOR, BCORL1, BRAF, CALR, CBL, CBLB, CBLC,CDKN2A, CEBPA, CSF3R, CUX1, DNMT3A, ETV6/TEL, EZH2, FBXW7, FLT3, GATA1, GATA2, GNAS, HRAS, IDH1, IDH2, IKZF1,JAK2, JAK3, KDM6A, KIT, KRAS, MLL, MPL, MYD88, NOTCH1, NPM1, NRAS, PDGFRA, PHF6, PTEN, PTPN11, RAD21, RUNX1,SETBP1, SF3B1, SMC1A, SMC3, SRSF2, STAG2, TET2, TP53, U2AF1, WT1, ZRSR2 Clinical Utility: This test is useful for the assessment of prognosis and/or treatment options for cancer patients at the initial diagnosis or at the relapse. -
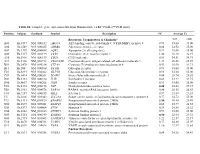
Table S1. Complete Gene Expression Data from Human Diabetes RT² Profiler™ PCR Array Receptors, Transporters & Channels* A
Table S1. Complete gene expression data from Human Diabetes RT² Profiler™ PCR Array Position Unigene GenBank Symbol Description FC Average Ct Receptors, Transporters & Channels* NGT GDM A01 Hs,5447 NM_000352 ABCC8 ATP-binding cassette, sub-family C (CFTR/MRP), member 8 0.93 35.00 35.00 A04 0Hs,2549 NM_000025 ADRB3 Adrenergic, beta-3-, receptor 0.88 34.92 35.00 A07 Hs,1307 NM_000486 AQP2 Aquaporin 2 (collecting duct) 0.93 35.00 35.00 A09 30Hs,5117 NM_001123 CCR2 Chemokine (C-C motif) receptor 2 1.00 26.28 26.17 A10 94Hs,5916 396NM_006139 CD28 CD28 molecule 0.81 34.51 34.71 A11 29Hs,5126 NM_001712 CEACAM1 Carcinoembryonic antigen-related cell adhesion molecule 1 1.31 26.08 25.59 B01 82Hs,2478 NM_005214 CTLA4 (biliaryCytotoxic glycoprotein) T-lymphocyte -associated protein 4 0.53 30.90 31.71 B11 24Hs,208 NM_000160 GCGR Glucagon receptor 0.93 35.00 35.00 C01 Hs,3891 NM_002062 GLP1R Glucagon-like peptide 1 receptor 0.93 35.00 35.00 C07 03Hs,6434 NM_000201 ICAM1 Intercellular adhesion molecule 1 0.84 28.74 28.89 D02 47Hs,5134 NM_000418 IL4R Interleukin 4 receptor 0.64 34.22 34.75 D06 57Hs,4657 NM_000208 INSR Insulin receptor 0.93 35.00 35.00 E05 44Hs,4312 NM_006178 NSF N-ethylmaleimide-sensitive factor 0.48 28.42 29.37 F08 79Hs,2961 NM_004578 RAB4A RAB4A, member RAS oncogene family 0.88 20.55 20.63 F10 69Hs,7287 NM_000655 SELL Selectin L 0.97 23.89 23.83 F11 56Hs,3806 NM_001042 SLC2A4 Solute carrier family 2 (facilitated glucose transporter), member 4 0.77 34.72 35.00 F12 91Hs,5111 NM_003825 SNAP23 Synaptosomal-associated protein, 23kDa 3.90 -

Genetic Testing for Acute Myeloid Leukemia AHS-M2062
Corporate Medical Policy Genetic Testing for Acute Myeloid Leukemia AHS-M2062 File Name: genetic_testing_for_acute_myeloid_leukemia Origination: 1/1/2019 Last CAP Review: 8/2021 Next CAP Review: 8/2022 Last Review: 8/2021 Description of Procedure or Service Acute myeloid leukemia (AML) is characterized by large numbers of abnormal, immature myeloid cells in the bone marrow and peripheral blood resulting from genetic changes in hematopoietic precursor cells which disrupt normal hematopoietic growth and differentiation (Stock, 2020). Related Policies: Genetic Cancer Susceptibility Using Next Generation Sequencing AHS-M2066 Molecular Panel Testing of Cancers to Identify Targeted Therapy AHS-M2109 Serum Tumor Markers for Malignancies AHS-G2124 Minimal Residual Disease (MRD) AHS- M2175 ***Note: This Medical Policy is complex and technical. For questions concerning the technical language and/or specific clinical indications for its use, please consult your physician. Policy BCBSNC will provide coverage for genetic testing for acute myeloid leukemia when it is determined to be medically necessary because the medical criteria and guidelines shown below are met. Benefits Application This medical policy relates only to the services or supplies described herein. Please refer to the Member's Benefit Booklet for availability of benefits. Member's benefits may vary according to benefit design; therefore member benefit language should be reviewed before applying the terms of this medical policy. When Genetic Testing for Acute Myeloid Leukemia is covered The use of genetic testing for acute myeloid leukemia is considered medically necessary for the following: A. Genetic testing for FLT3 internal tandem duplication and tyrosine kinase domain mutations (ITD and TKD), IDH1, IDH2, TET2, WT1, DNMT3A, ASXL1 and/or TP53 in adult and pediatric patients with suspected or confirmed AML of any type for prognostic and/or therapeutic purposes. -

Regulation of the C/Ebpα Signaling Pathway in Acute Myeloid Leukemia (Review)
ONCOLOGY REPORTS 33: 2099-2106, 2015 Regulation of the C/EBPα signaling pathway in acute myeloid leukemia (Review) GUANHUA SONG1, LIn Wang2, Kehong BI3 and guosheng JIang1 1Department of hemato-oncology, Institute of Basic Medicine, shandong academy of Medical sciences, Key Laboratory for Modern Medicine and Technology of Shandong Province, Key Laboratory for Rare and Uncommon Diseases, Key Medical Laboratory for Tumor Immunology and Traditional Chinese Medicine Immunology of shandong Province, Jinan, Shandong 250062; 2Research Center for Medical Biotechnology, Shandong Academy of Medical Sciences, Jinan, Shandong 250062; 3Department of Hematology, Qianfoshan Mountain Hospital of Shandong University, Jinan, Shandong 250014, P.R. China Received December 2, 2014; Accepted January 26, 2015 DoI: 10.3892/or.2015.3848 Abstract. The transcription factor CCAAT/enhancer binding Contents protein α (C/EBPα), as a critical regulator of myeloid devel- opment, directs granulocyte and monocyte differentiation. 1. Introduction Various mechanisms have been identified to explain how 2. Function of C/EBPα in myeloid differentiation C/EBPα functions in patients with acute myeloid leukemia 3. Regulation of the C/EBPα signaling pathway (AML). C/EBPα expression is suppressed as a result of 4. Conclusion common leukemia-associated genetic and epigenetic altera- tions such as AML1-ETO, RARα-PLZF or gene promoter methylation. Recent data have shown that ubiquitination modi- 1. Introduction fication also contributes to its downregulation. In addition, 10-15% of patients with AML in an intermediate cytogenetic Acute myeloid leukemia (AML) is characterized by uncon- risk subgroup were characterized by mutations of the C/EBPα trolled proliferation of myeloid progenitors that exhibit a gene. -

CEBPA Gene CCAAT Enhancer Binding Protein Alpha
CEBPA gene CCAAT enhancer binding protein alpha Normal Function The CEBPA gene provides instructions for making a protein called CCAAT enhancer- binding protein alpha. This protein is a transcription factor, which means that it attaches ( binds) to specific regions of DNA and helps control the activity (expression) of certain genes. CCAAT enhancer-binding protein alpha is involved in the maturation ( differentiation) of certain blood cells. It is also believed to act as a tumor suppressor, which means that it is involved in cellular mechanisms that help prevent the cells from growing and dividing too rapidly or in an uncontrolled way. Health Conditions Related to Genetic Changes Familial acute myeloid leukemia with mutated CEBPA At least six mutations in the CEBPA gene have been identified in families with familial acute myeloid leukemia with mutated CEBPA, which is a form of a blood cancer known as acute myeloid leukemia. These inherited mutations are present throughout a person' s life in virtually every cell in the body. The mutations result in a shorter version of CCAAT enhancer-binding protein alpha. This shortened protein is produced from one copy of the CEBPA gene in each cell, and it is believed to interfere with the tumor suppressor function of the normal protein produced from the second copy of the gene. Absence of the tumor suppressor function of CCAAT enhancer-binding protein alpha is believed to disrupt the regulation of blood cell production, leading to the uncontrolled production of abnormal cells that occurs in acute myeloid leukemia. In addition to the inherited mutation in one copy of the CEBPA gene in each cell, most individuals with familial acute myeloid leukemia with mutated CEBPA also acquire a mutation in the second copy of the CEBPA gene. -

The Polymorphism Rs944289 Predisposes to Papillary Thyroid Carcinoma Through a Large Intergenic Noncoding RNA Gene of Tumor Suppressor Type
The polymorphism rs944289 predisposes to papillary thyroid carcinoma through a large intergenic noncoding RNA gene of tumor suppressor type Jaroslaw Jendrzejewskia, Huiling Hea, Hanna S. Radomskaa, Wei Lia, Jerneja Tomsica, Sandya Liyanarachchia, Ramana V. Davulurib, Rebecca Nagya, and Albert de la Chapellea,1 aHuman Cancer Genetics Program, Comprehensive Cancer Center, The Ohio State University, Columbus, OH 43210; and bMolecular and Cellular Oncogenesis Program, Center for Systems and Computational Biology, The Wistar Institute, Philadelphia, PA 19104 Contributed by Albert de la Chapelle, April 4, 2012 (sent for review February 20, 2012) A genome-wide association study of papillary thyroid carcinoma (GWAS) have addressed the predisposition to PTC (11–13). (PTC) pinpointed two independent SNPs (rs944289 and rs965513) Gudmundsson et al. (11) studied Icelandic PTC patients and located in regions containing no annotated genes (14q13.3 and controls, followed by validation in Spanish and US cases and 9q22.33, respectively). Here, we describe a unique, long, inter- controls. Two SNPs that showed highly significant association Papillary Thyroid genic, noncoding RNA gene (lincRNA) named with PTC were detected (rs965513 and rs944289). The variants Carcinoma Susceptibility Candidate 3 PTCSC3 ( ) located 3.2 kb were located in 9q22.33 and 14q13.3, respectively, and have been downstream of rs944289 at 14q.13.3 and the expression of which confirmed by other research groups (14, 15). As both SNPs were is strictly thyroid specific. By quantitative PCR, PTCSC3 expression − located in gene-poor regions, no immediate candidate genes was strongly down-regulated (P = 2.84 × 10 14) in thyroid tumor tissue of 46 PTC patients and the risk allele (T) was associated with presented themselves as being affected by or associated with the the strongest suppression (genotype [TT] (n = 21) vs. -

Identification of Microrna-Mrna Regulatory Networks and Pathways Related to Retinoblastoma Across Human and Mouse
Int J Ophthalmol, Vol. 13, No. 4, Apr.18, 2020 www.ijo.cn Tel: 8629-82245172 8629-82210956 Email: [email protected] ·Basic Research· Identification of microRNA-mRNA regulatory networks and pathways related to retinoblastoma across human and mouse Rui Tian, He Zou, Lu-Fei Wang, Mei-Jiao Song, Lu Liu, Hui Zhang Department of Ophthalmology, the Second Hospital of Jilin INTRODUCTION University, Changchun 130000, Jilin Province, China etinoblastoma (RB) is a rare malignant retina tumor Correspondence to: Hui Zhang. Department of Ophthalmology, R in children. It initiates during foetal stage and is often the Second Hospital of Jilin University, No.218 Ziqiang Street, delay-diagnosed after birth or during the first few years Nan Guan District, Changchun 130000, Jilin Province, China. after birth. The common signs of RB includes leukocoria, [email protected] strabismus, glaucoma and inflammation[1-2]. The prognosis and Received: 2019-07-20 Accepted: 2020-02-19 survival of RB has been improved due to the improvement in diagnosis methods and treatment strategies, including Abstract chemotherapy regiments, magnetic resonance imaging and ● AIM: To explore the mRNA and pathways related to surgical therapy[1-3]. However, the etiology of RB is still now retinoblastoma (RB) genesis and development. clear till now. ● METHODS: Microarray datasets GSE29683 (human) The pathogenesis of RB involves RB tumor suppressor and GSE29685 (mouse) were downloaded from NCBI GEO gene biallelic mutation or inactivation and attendant loss of database. Homologous genes between the two species function of RB protein[1]. It is believed that the molecular were identified using WGCNA, followed by protein-protein features of a disease reflect its origin features and provide interaction (PPI) network construction and gene enrichment clues for treatment. -

Global Analysis of Estrogen Receptor Beta Binding to Breast Cancer Cell Genome Reveals an Extensive Interplay with Estrogen Rece
Grober et al. BMC Genomics 2011, 12:36 http://www.biomedcentral.com/1471-2164/12/36 RESEARCHARTICLE Open Access Global analysis of estrogen receptor beta binding to breast cancer cell genome reveals an extensive interplay with estrogen receptor alpha for target gene regulation Oli MV Grober1†, Margherita Mutarelli1†, Giorgio Giurato1, Maria Ravo1,2, Luigi Cicatiello1, Maria Rosaria De Filippo1, Lorenzo Ferraro1, Giovanni Nassa1,2, Maria Francesca Papa1, Ornella Paris1, Roberta Tarallo1,2, Shujun Luo3, Gary P Schroth3, Vladimir Benes4, Alessandro Weisz1,2* Abstract Background: Estrogen receptors alpha (ERa) and beta (ERb) are transcription factors (TFs) that mediate estrogen signaling and define the hormone-responsive phenotype of breast cancer (BC). The two receptors can be found co-expressed and play specific, often opposite, roles, with ERb being able to modulate the effects of ERa on gene transcription and cell proliferation. ERb is frequently lost in BC, where its presence generally correlates with a better prognosis of the disease. The identification of the genomic targets of ERb in hormone-responsive BC cells is thus a critical step to elucidate the roles of this receptor in estrogen signaling and tumor cell biology. Results: Expression of full-length ERb in hormone-responsive, ERa-positive MCF-7 cells resulted in a marked reduction in cell proliferation in response to estrogen and marked effects on the cell transcriptome. By ChIP-Seq we identified 9702 ERb and 6024 ERa binding sites in estrogen-stimulated cells, comprising sites occupied by either ERb,ERa or both ER subtypes. A search for TF binding matrices revealed that the majority of the binding sites identified comprise one or more Estrogen Response Element and the remaining show binding matrixes for other TFs known to mediate ER interaction with chromatin by tethering, including AP2, E2F and SP1. -

Acute Myeloid Leukemia with Biallelic CEBPA Gene Mutations and Normal Karyotype Represents a Distinct Genetic Entity Associated
VOLUME 28 ⅐ NUMBER 4 ⅐ FEBRUARY 1 2010 JOURNAL OF CLINICAL ONCOLOGY ORIGINAL REPORT Acute Myeloid Leukemia With Biallelic CEBPA Gene Mutations and Normal Karyotype Represents a Distinct Genetic Entity Associated With a Favorable Clinical Outcome Annika Dufour, Friederike Schneider, Klaus H. Metzeler, Eva Hoster, Stephanie Schneider, Evelyn Zellmeier, Tobias Benthaus, Maria-Cristina Sauerland, Wolfgang E. Berdel, Thomas Bu¨chner, Bernhard Wo¨rmann, Jan Braess, Wolfgang Hiddemann, Stefan K. Bohlander, and Karsten Spiekermann See accompanying article on page 578 From the Laboratory for Leukemia Diag- nostics, Department of Internal Medi- ABSTRACT cine III; Institute for Medical Informatics, Biometry and Epidemiol- Purpose ogy, University of Munich-Grosshadern; CEBPA mutations are found as either biallelic (biCEBPA) or monoallelic (moCEBPA). We set out to Clinical Cooperative Group “Leukemia,” explore whether the kind of CEBPA mutation is of prognostic relevance in cytogenetically normal HelmholtzZentrum Mu¨nchen, German (CN) acute myeloid leukemia (AML). Research Center for Environmental Health, Munich; Departments of Medi- Patients and Methods cal Informatics and Biomathematics and Four hundred sixty-seven homogeneously treated patients with CN-AML were subdivided into Medicine A, Hematology and Oncology, moCEBPA,biCEBPA, and wild-type (wt) CEBPA patients. The subgroups were analyzed for clinical University of Mu¨nster, Mu¨nster; and parameters and for additional mutations in the NPM1, FLT3, and MLL genes. Furthermore, we Department of Hematology and Oncol- obtained gene expression profiles using oligonucleotide microarrays. ogy, Municipal Hospital, Braunschweig, Germany. Results Only patients with biCEBPA had an improved median overall survival when compared with Submitted December 22, 2008; ϭ accepted June 4, 2009; published patients with wtCEBPA (not reached v 20.4 months, respectively; P .018), whereas patients online ahead of print at www.jco.org on with moCEBPA (20.9 months) and wtCEBPA had a similar outcome (P ϭ .506). -

In Silico Genetics Revealing Novel Mutations in CEBPA Gene Associated with Acute Myeloid Leukemia
bioRxiv preprint doi: https://doi.org/10.1101/608943; this version posted April 14, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. In Silico Genetics Revealing Novel Mutations in CEBPA Gene Associated with Acute Myeloid Leukemia Mujahed I. Mustafa1, Zainab O. Mohammed2, Naseem S. Murshed1, Nafisa M. Elfadol1, Abdelrahman H. Abdelmoneim1, Mohamed A. Hassan1 1-Department of Biotechnology, Africa city of Technology, Sudan. 2- Hematology Department, Ribat University Hospital, Sudan. *Corresponding Author: Mujahed I. Mustafa: [email protected] Abstract: Background: Myelodysplastic syndrome/Acute myeloid leukemia (MDS/AML) is a highly heterogeneous malignant disease; affects children and adults of all ages. AML is one of the main causes of death in children with cancer. However, It is the most common acute leukemia in adults, with a frequency of over 20 000 cases per year in the United States of America alone. Methods: The SNPs were retrieved from the dbSNP database. this SNPs were submitted into various functional analysis tools that done by SIFT, PolyPhen-2, PROVEAN, SNAP2, SNPs&GO, PhD-SNP and PANTHER, while structural analysis were done by I-mutant3 and MUPro. The most damaging SNPs were selected for further analysis by Mutation3D, Project hope, ConSurf and BioEdit softwares. Results: A total of five novel nsSNPs out of 248 missense mutations were predicted to be responsible for the structural and functional variations of CEBPA protein. -

Mirnas and Pirnas from Bone Marrow Mesenchymal Stem Cell Extracellular Vesicles Induce Cell Survival and Inhibit Cell Differenti
www.impactjournals.com/oncotarget/ Oncotarget, Vol. 7, No. 6 MiRNAs and piRNAs from bone marrow mesenchymal stem cell extracellular vesicles induce cell survival and inhibit cell differentiation of cord blood hematopoietic stem cells: a new insight in transplantation Luciana De Luca1, Stefania Trino1, Ilaria Laurenzana1, Vittorio Simeon1, Giovanni Calice1, Stefania Raimondo2, Marina Podestà3, Michele Santodirocco4, Lazzaro Di Mauro4, Francesco La Rocca1, Antonella Caivano1, Annalisa Morano1, Francesco Frassoni5, Daniela Cilloni2, Luigi Del Vecchio6,7,*, Pellegrino Musto8,* 1 Laboratory of Preclinical and Translational Research, IRCCS-Centro di Riferimento Oncologico della Basilicata (CROB), Rionero in Vulture, 85028, Italy 2Department of Clinical and Biological Sciences, University of Turin, Turin 10126, Italy 3Stem Cell Center, S. Martino Hospital, Genova 16132, Italy 4 Transfusion Medicine Unit, Puglia Cord Blood Bank, IRCCS-Casa Sollievo della Sofferenza, San Giovanni Rotondo, 71013, Italy 5Laboratorio Cellule Staminali post natali e Terapie Cellulari, Giannina Gaslini Institute, Genova 16148, Italy 6 CEINGE-Biotecnologie Avanzate S.C.a R.L., Naples, 80145, Italy 7Department of Molecular Medicine and Medical Biotechnologies, Federico II University, Naples 80131, Italy 8Scientific Direction, IRCCS-Centro di Riferimento Oncologico Basilicata (CROB), Rionero in Vulture, 85028, Italy *These authors contributed equally to this work Correspondence to: Luciana De Luca, e-mail: [email protected] Keywords: mesenchymal stem cells, extracellular vesicles, umbilical cord blood stem cells, microRNAs, piRNAs Received: June 15, 2015 Accepted: December 05, 2015 Published: December 29, 2015 ABSTRACT Hematopoietic stem cells (HSC), including umbilical cord blood CD34+ stem cells (UCB-CD34+), are used for the treatment of several diseases. Although different studies suggest that bone marrow mesenchymal stem cells (BM-MSC) support hematopoiesis, the exact mechanism remains unclear.