Overexpression of the ETS-Related Gene
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Brazilian Cohort of Individuals with Phelan-Mcdermid Syndrome
Samogy-Costa et al. Journal of Neurodevelopmental Disorders (2019) 11:13 https://doi.org/10.1186/s11689-019-9273-1 RESEARCH Open Access A Brazilian cohort of individuals with Phelan-McDermid syndrome: genotype- phenotype correlation and identification of an atypical case Claudia Ismania Samogy-Costa1†, Elisa Varella-Branco1†, Frederico Monfardini1, Helen Ferraz2, Rodrigo Ambrósio Fock3, Ricardo Henrique Almeida Barbosa3, André Luiz Santos Pessoa4,5, Ana Beatriz Alvarez Perez3, Naila Lourenço1, Maria Vibranovski1, Ana Krepischi1, Carla Rosenberg1 and Maria Rita Passos-Bueno1* Abstract Background: Phelan-McDermid syndrome (PMS) is a rare genetic disorder characterized by global developmental delay, intellectual disability (ID), autism spectrum disorder (ASD), and mild dysmorphisms associated with several comorbidities caused by SHANK3 loss-of-function mutations. Although SHANK3 haploinsufficiency has been associated with the major neurological symptoms of PMS, it cannot explain the clinical variability seen among individuals. Our goals were to characterize a Brazilian cohort of PMS individuals, explore the genotype-phenotype correlation underlying this syndrome, and describe an atypical individual with mild phenotype. Methodology: A total of 34 PMS individuals were clinically and genetically evaluated. Data were obtained by a questionnaire answered by parents, and dysmorphic features were assessed via photographic evaluation. We analyzed 22q13.3 deletions and other potentially pathogenic copy number variants (CNVs) and also performed genotype-phenotype correlation analysis to determine whether comorbidities, speech status, and ASD correlate to deletion size. Finally, a Brazilian cohort of 829 ASD individuals and another independent cohort of 2297 ID individuals was used to determine the frequency of PMS in these disorders. Results: Our data showed that 21% (6/29) of the PMS individuals presented an additional rare CNV, which may contribute to clinical variability in PMS. -

A Randomized Controlled Trial of Intranasal Oxytocin in Phelan-Mcdermid Syndrome
A Randomized Controlled Trial of Intranasal Oxytocin in Phelan-McDermid Syndrome Jarrett Fastman Icahn School of Medicine at Mount Sinai Jennifer Foss-Feig Icahn School of Medicine at Mount Sinai Yitzchak Frank Icahn School of Medicine at Mount Sinai Danielle Halpern Icahn School of Medicine at Mount Sinai Hala Harony-Nicolas Icahn School of Medicine at Mount Sinai Christina Layton Icahn School of Medicine at Mount Sinai Sven Sandin Icahn School of Medicine at Mount Sinai Paige Siper Icahn School of Medicine at Mount Sinai Lara Tang Icahn School of Medicine at Mount Sinai Pilar Trelles Icahn School of Medicine at Mount Sinai Jessica Zweifach Icahn School of Medicine at Mount Sinai Joseph D. Buxbaum Icahn School of Medicine at Mount Sinai Alexander Kolevzon ( [email protected] ) Icahn School of Medicine at Mount Sinai https://orcid.org/0000-0001-8129-2671 Research Article Keywords: Phelan-McDermid syndrome, PMS, shank3, autism spectrum disorder, ASD, oxytocin Posted Date: March 5th, 2021 Page 1/24 DOI: https://doi.org/10.21203/rs.3.rs-268151/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 2/24 Abstract Background Phelan-McDermid syndrome (PMS) is a rare neurodevelopmental disorder caused by haploinsuciency of the SHANK3 gene and characterized by global developmental delays, decits in speech and motor function, and autism spectrum disorder (ASD). Monogenic causes of ASD such as PMS are well suited to investigations with novel therapeutics, as interventions can be targeted based on established genetic etiology. While preclinical studies have demonstrated that the neuropeptide oxytocin can reverse electrophysiological, attentional, and social recognition memory decits in Shank3-decient rats, there have been no trials in individuals with PMS. -

22Q13.3 Deletion Syndrome
22q13.3 deletion syndrome Description 22q13.3 deletion syndrome, which is also known as Phelan-McDermid syndrome, is a disorder caused by the loss of a small piece of chromosome 22. The deletion occurs near the end of the chromosome at a location designated q13.3. The features of 22q13.3 deletion syndrome vary widely and involve many parts of the body. Characteristic signs and symptoms include developmental delay, moderate to profound intellectual disability, decreased muscle tone (hypotonia), and absent or delayed speech. Some people with this condition have autism or autistic-like behavior that affects communication and social interaction, such as poor eye contact, sensitivity to touch, and aggressive behaviors. They may also chew on non-food items such as clothing. Less frequently, people with this condition have seizures or lose skills they had already acquired (developmental regression). Individuals with 22q13.3 deletion syndrome tend to have a decreased sensitivity to pain. Many also have a reduced ability to sweat, which can lead to a greater risk of overheating and dehydration. Some people with this condition have episodes of frequent vomiting and nausea (cyclic vomiting) and backflow of stomach acids into the esophagus (gastroesophageal reflux). People with 22q13.3 deletion syndrome typically have distinctive facial features, including a long, narrow head; prominent ears; a pointed chin; droopy eyelids (ptosis); and deep-set eyes. Other physical features seen with this condition include large and fleshy hands and/or feet, a fusion of the second and third toes (syndactyly), and small or abnormal toenails. Some affected individuals have rapid (accelerated) growth. -

C/Ebpα Is an Essential Collaborator in Hoxa9/Meis1-Mediated Leukemogenesis
C/EBPα is an essential collaborator in Hoxa9/Meis1-mediated leukemogenesis Cailin Collinsa, Jingya Wanga, Hongzhi Miaoa, Joel Bronsteina, Humaira Nawera, Tao Xua, Maria Figueroaa, Andrew G. Munteana, and Jay L. Hessa,b,1 aDepartment of Pathology, University of Michigan, Ann Arbor, MI 48109; and bIndiana University School of Medicine, Indianapolis, IN 46202 Edited* by Louis M. Staudt, National Institutes of Health, Bethesda, MD, and approved May 19, 2014 (received for review February 12, 2014) Homeobox A9 (HOXA9) is a homeodomain-containing transcrip- with Hoxa9. In addition, C/EBP recognition motifs are enriched tion factor that plays a key role in hematopoietic stem cell expan- at Hoxa9 binding sites. sion and is commonly deregulated in human acute leukemias. A C/EBPα is a basic leucine-zipper transcription factor that plays variety of upstream genetic alterations in acute myeloid leukemia a critical role in lineage commitment during hematopoietic dif- −/− (AML) lead to overexpression of HOXA9, almost always in associ- ferentiation (18). Whereas Cebpa mice show complete loss of ation with overexpression of its cofactor meis homeobox 1 (MEIS1). the granulocytic compartment, recent work shows that loss of α A wide range of data suggests that HOXA9 and MEIS1 play a syn- C/EBP in adult HSCs leads to both an increase in the number ergistic causative role in AML, although the molecular mechanisms of functional HSCs and an increase in their proliferative and leading to transformation by HOXA9 and MEIS1 remain elusive. In repopulating capacity (19, 20). Conversely, CEBPA overexpression can promote transdifferentiation of a variety of fibroblastic cells to this study, we identify CCAAT/enhancer binding protein alpha (C/ the myeloid lineage and can induce monocytic differentiation in EBPα) as a critical collaborator required for Hoxa9/Meis1-mediated α MLL-fusion protein-mediated leukemias (21, 22). -

Test Summary Flyer-NGS Panels.Pub
Cancer‐related Mutaon Analysis Next Generaon Gene Sequencing for Myeloid Tesng Assay Summary IU Health Molecular Pathology Laboratory now offers high throughput sequencing for hot spot mutations found in clinically relevant cancer genes. In addition to a general panel of 54 genes, selected panels have been developed for a more tai- lored application in specific cancers. Comparing to single gene assay, these panels offer a more comprehensive and eco- nomic way to assess prognosis and/or treatment options for cancer patients at the initial diagnosis or at the relapse. Orderable Name: Use IU Health Molecular Pathology requisition; Call 317.491.6417 for requisition. Panels include: AML Mutations by NGS ASXL1, CEBPA, DNMT3A, ETV6/TEL, FLT3, HRAS, IDH1, IDH2, KIT, KRAS, MLL, NPM1, NRAS, PHF6, RUNX1, TET2, TP53, WT1 MDS Mutations by NGS ASXL1, ATRX, BCOR, BCORL1, ETV6/TEL, DNMT3A, EZH2, GNAS, IDH1, IDH2, RUNX1, SF3B1, SRSF2, TET2, TP53, U2AF1, ZRSR2 CML Mutations by NGS ABL1 MPN Mutations by NGS ASXL1, BRAF, CALR, CSF3R, EZH2, IKZF1, JAK2, JAK3, KDM6A, KIT, MPL, PDGRA, SETBP1, TET2 CMML Mutations by NGS ASXL1, CBL, CBLB, CBLC, EZH2, RUNX1, TET2, TP53, SRSF2 JMML Mutations by NGS CBL, CBLB, CBLC, HRAS, KRAS, NRAS, PTPN11 ALL Mutations by NGS ABL1, CSF3R, FBXW7, IKZF1, JAK3, KDM6A, NOTCH1 CLL Mutations by NGS MYD88, NOTCH1, SF3B1, TP53 Lymphoma/Myeloma Mutations by NGS BRAF, CDKN2A, CSF3R, FBXW7, HRAS, KRAS, MYD88, NOTCH1, NRAS, SF3B1,TP53 Hematopoietic Neoplasms Mutations by NGS ABL1, ASXL1, ATRX, BCOR, BCORL1, BRAF, CALR, CBL, CBLB, CBLC,CDKN2A, CEBPA, CSF3R, CUX1, DNMT3A, ETV6/TEL, EZH2, FBXW7, FLT3, GATA1, GATA2, GNAS, HRAS, IDH1, IDH2, IKZF1,JAK2, JAK3, KDM6A, KIT, KRAS, MLL, MPL, MYD88, NOTCH1, NPM1, NRAS, PDGFRA, PHF6, PTEN, PTPN11, RAD21, RUNX1,SETBP1, SF3B1, SMC1A, SMC3, SRSF2, STAG2, TET2, TP53, U2AF1, WT1, ZRSR2 Clinical Utility: This test is useful for the assessment of prognosis and/or treatment options for cancer patients at the initial diagnosis or at the relapse. -
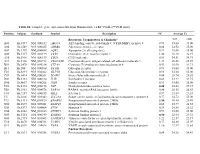
Table S1. Complete Gene Expression Data from Human Diabetes RT² Profiler™ PCR Array Receptors, Transporters & Channels* A
Table S1. Complete gene expression data from Human Diabetes RT² Profiler™ PCR Array Position Unigene GenBank Symbol Description FC Average Ct Receptors, Transporters & Channels* NGT GDM A01 Hs,5447 NM_000352 ABCC8 ATP-binding cassette, sub-family C (CFTR/MRP), member 8 0.93 35.00 35.00 A04 0Hs,2549 NM_000025 ADRB3 Adrenergic, beta-3-, receptor 0.88 34.92 35.00 A07 Hs,1307 NM_000486 AQP2 Aquaporin 2 (collecting duct) 0.93 35.00 35.00 A09 30Hs,5117 NM_001123 CCR2 Chemokine (C-C motif) receptor 2 1.00 26.28 26.17 A10 94Hs,5916 396NM_006139 CD28 CD28 molecule 0.81 34.51 34.71 A11 29Hs,5126 NM_001712 CEACAM1 Carcinoembryonic antigen-related cell adhesion molecule 1 1.31 26.08 25.59 B01 82Hs,2478 NM_005214 CTLA4 (biliaryCytotoxic glycoprotein) T-lymphocyte -associated protein 4 0.53 30.90 31.71 B11 24Hs,208 NM_000160 GCGR Glucagon receptor 0.93 35.00 35.00 C01 Hs,3891 NM_002062 GLP1R Glucagon-like peptide 1 receptor 0.93 35.00 35.00 C07 03Hs,6434 NM_000201 ICAM1 Intercellular adhesion molecule 1 0.84 28.74 28.89 D02 47Hs,5134 NM_000418 IL4R Interleukin 4 receptor 0.64 34.22 34.75 D06 57Hs,4657 NM_000208 INSR Insulin receptor 0.93 35.00 35.00 E05 44Hs,4312 NM_006178 NSF N-ethylmaleimide-sensitive factor 0.48 28.42 29.37 F08 79Hs,2961 NM_004578 RAB4A RAB4A, member RAS oncogene family 0.88 20.55 20.63 F10 69Hs,7287 NM_000655 SELL Selectin L 0.97 23.89 23.83 F11 56Hs,3806 NM_001042 SLC2A4 Solute carrier family 2 (facilitated glucose transporter), member 4 0.77 34.72 35.00 F12 91Hs,5111 NM_003825 SNAP23 Synaptosomal-associated protein, 23kDa 3.90 -

Three-Dimensional Fluorescence in Situ Hybridization in Mouse
Three-Dimensional Fluorescence In Situ Hybridization in Mouse Embryos Using Repetitive Probe Sequences Walid Maalouf, Tiphaine Aguirre-Lavin, Laetitia Herzog, Isabelle Bataillon, Pascale Debey, Nathalie Beaujean To cite this version: Walid Maalouf, Tiphaine Aguirre-Lavin, Laetitia Herzog, Isabelle Bataillon, Pascale Debey, et al.. Three-Dimensional Fluorescence In Situ Hybridization in Mouse Embryos Using Repetitive Probe Sequences. Fluorescence in situ Hybridization (FISH), 659 (4), Springer, pp.401-408, 2010, Methods in Molecular Biology, 10.1007/978-1-60761-789-1_31. hal-02610638 HAL Id: hal-02610638 https://hal.archives-ouvertes.fr/hal-02610638 Submitted on 17 May 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. 1 Three-Dimensional Fluorescent In Situ Hybridisation in Mouse Embryos Walid E. Maalouf1,2, Tiphaine Aguirre-Lavin1, Laetitia Herzog1, Isabelle Bataillon1, Pascale Debey1 and Nathalie Beaujean1 1INRA, UMR 1198 Biologie du Développement et Reproduction, F-78350 Jouy en Josas, France 32 Present Address: QMRI, 47 Little France Crescent, University of Edinburgh, Edinburgh, UK Contact : Dr Walid Maalouf <[email protected]>, Tel. +33 (0)1 34 65 29 03 / Fax: 29 09 Abstract A common problem in research laboratories that study the mammalian embryo is the limited supply of live material. -

Genetic Testing for Acute Myeloid Leukemia AHS-M2062
Corporate Medical Policy Genetic Testing for Acute Myeloid Leukemia AHS-M2062 File Name: genetic_testing_for_acute_myeloid_leukemia Origination: 1/1/2019 Last CAP Review: 8/2021 Next CAP Review: 8/2022 Last Review: 8/2021 Description of Procedure or Service Acute myeloid leukemia (AML) is characterized by large numbers of abnormal, immature myeloid cells in the bone marrow and peripheral blood resulting from genetic changes in hematopoietic precursor cells which disrupt normal hematopoietic growth and differentiation (Stock, 2020). Related Policies: Genetic Cancer Susceptibility Using Next Generation Sequencing AHS-M2066 Molecular Panel Testing of Cancers to Identify Targeted Therapy AHS-M2109 Serum Tumor Markers for Malignancies AHS-G2124 Minimal Residual Disease (MRD) AHS- M2175 ***Note: This Medical Policy is complex and technical. For questions concerning the technical language and/or specific clinical indications for its use, please consult your physician. Policy BCBSNC will provide coverage for genetic testing for acute myeloid leukemia when it is determined to be medically necessary because the medical criteria and guidelines shown below are met. Benefits Application This medical policy relates only to the services or supplies described herein. Please refer to the Member's Benefit Booklet for availability of benefits. Member's benefits may vary according to benefit design; therefore member benefit language should be reviewed before applying the terms of this medical policy. When Genetic Testing for Acute Myeloid Leukemia is covered The use of genetic testing for acute myeloid leukemia is considered medically necessary for the following: A. Genetic testing for FLT3 internal tandem duplication and tyrosine kinase domain mutations (ITD and TKD), IDH1, IDH2, TET2, WT1, DNMT3A, ASXL1 and/or TP53 in adult and pediatric patients with suspected or confirmed AML of any type for prognostic and/or therapeutic purposes. -

Regulation of the C/Ebpα Signaling Pathway in Acute Myeloid Leukemia (Review)
ONCOLOGY REPORTS 33: 2099-2106, 2015 Regulation of the C/EBPα signaling pathway in acute myeloid leukemia (Review) GUANHUA SONG1, LIn Wang2, Kehong BI3 and guosheng JIang1 1Department of hemato-oncology, Institute of Basic Medicine, shandong academy of Medical sciences, Key Laboratory for Modern Medicine and Technology of Shandong Province, Key Laboratory for Rare and Uncommon Diseases, Key Medical Laboratory for Tumor Immunology and Traditional Chinese Medicine Immunology of shandong Province, Jinan, Shandong 250062; 2Research Center for Medical Biotechnology, Shandong Academy of Medical Sciences, Jinan, Shandong 250062; 3Department of Hematology, Qianfoshan Mountain Hospital of Shandong University, Jinan, Shandong 250014, P.R. China Received December 2, 2014; Accepted January 26, 2015 DoI: 10.3892/or.2015.3848 Abstract. The transcription factor CCAAT/enhancer binding Contents protein α (C/EBPα), as a critical regulator of myeloid devel- opment, directs granulocyte and monocyte differentiation. 1. Introduction Various mechanisms have been identified to explain how 2. Function of C/EBPα in myeloid differentiation C/EBPα functions in patients with acute myeloid leukemia 3. Regulation of the C/EBPα signaling pathway (AML). C/EBPα expression is suppressed as a result of 4. Conclusion common leukemia-associated genetic and epigenetic altera- tions such as AML1-ETO, RARα-PLZF or gene promoter methylation. Recent data have shown that ubiquitination modi- 1. Introduction fication also contributes to its downregulation. In addition, 10-15% of patients with AML in an intermediate cytogenetic Acute myeloid leukemia (AML) is characterized by uncon- risk subgroup were characterized by mutations of the C/EBPα trolled proliferation of myeloid progenitors that exhibit a gene. -

Advances in Autism Genetics: on the Threshold of a New Neurobiology
REVIEWS Advances in autism genetics: on the threshold of a new neurobiology Brett S. Abrahams and Daniel H. Geschwind Abstract | Autism is a heterogeneous syndrome defined by impairments in three core domains: social interaction, language and range of interests. Recent work has led to the identification of several autism susceptibility genes and an increased appreciation of the contribution of de novo and inherited copy number variation. Promising strategies are also being applied to identify common genetic risk variants. Systems biology approaches, including array-based expression profiling, are poised to provide additional insights into this group of disorders, in which heterogeneity, both genetic and phenotypic, is emerging as a dominant theme. Gene association studies Autistic disorder is the most severe end of a group of into the ASDs. This work, in concert with important A set of methods that is used neurodevelopmental disorders referred to as autism technical advances, made it possible to carry out the to determine the correlation spectrum disorders (ASDs), all of which share the com- first candidate gene association studies and resequenc- (positive or negative) between mon feature of dysfunctional reciprocal social interac- ing efforts in the late 1990s. Whole-genome linkage a defined genetic variant and a studies phenotype of interest. tion. A meta-analysis of ASD prevalence rates suggests followed, and were used to identify additional that approximately 37 in 10,000 individuals are affected1. loci of potential interest. Although -

CEBPA Gene CCAAT Enhancer Binding Protein Alpha
CEBPA gene CCAAT enhancer binding protein alpha Normal Function The CEBPA gene provides instructions for making a protein called CCAAT enhancer- binding protein alpha. This protein is a transcription factor, which means that it attaches ( binds) to specific regions of DNA and helps control the activity (expression) of certain genes. CCAAT enhancer-binding protein alpha is involved in the maturation ( differentiation) of certain blood cells. It is also believed to act as a tumor suppressor, which means that it is involved in cellular mechanisms that help prevent the cells from growing and dividing too rapidly or in an uncontrolled way. Health Conditions Related to Genetic Changes Familial acute myeloid leukemia with mutated CEBPA At least six mutations in the CEBPA gene have been identified in families with familial acute myeloid leukemia with mutated CEBPA, which is a form of a blood cancer known as acute myeloid leukemia. These inherited mutations are present throughout a person' s life in virtually every cell in the body. The mutations result in a shorter version of CCAAT enhancer-binding protein alpha. This shortened protein is produced from one copy of the CEBPA gene in each cell, and it is believed to interfere with the tumor suppressor function of the normal protein produced from the second copy of the gene. Absence of the tumor suppressor function of CCAAT enhancer-binding protein alpha is believed to disrupt the regulation of blood cell production, leading to the uncontrolled production of abnormal cells that occurs in acute myeloid leukemia. In addition to the inherited mutation in one copy of the CEBPA gene in each cell, most individuals with familial acute myeloid leukemia with mutated CEBPA also acquire a mutation in the second copy of the CEBPA gene. -

Cryptic Subtelomeric Translocations in the 22Q13 Deletion Syndrome J Med Genet: First Published As 10.1136/Jmg.37.1.58 on 1 January 2000
58 J Med Genet 2000;37:58–61 Cryptic subtelomeric translocations in the 22q13 deletion syndrome J Med Genet: first published as 10.1136/jmg.37.1.58 on 1 January 2000. Downloaded from Verayuth Praphanphoj, Barbara K Goodman, George H Thomas, Gerald V Raymond Abstract lieved to result from de novo, simple, subtelom- Cryptic subtelomeric rearrangements eric deletions,1–9 two cases were derived from are suspected to underlie a substantial balanced translocations,49 one case was the portion of terminal chromosomal dele- result of a familial chromosome 22 inversion,10 tions. We have previously described two and in one case the mechanism was not children, one with an unbalanced subtelo- determined.11 (It is possible that one of these meric rearrangement resulting in dele- cases may have been reported twice,17in which tion of 22q13→qter and duplication of case the total number would be 21 cases and the 1qter, and a second with an apparently simple deletions would be 17 cases.) This report simple 22q13→qter deletion. We have describes the clinical findings in a new case of examined two additional patients with 22q13→qter deletion and the identification of deletions of 22q13→qter. In one of the new translocated material on the deleted chromo- patients presented here, clinical findings some using multi-telomere fluorescence in situ were suggestive of the 22q13 deletion syn- hybridisation (FISH). drome and FISH for 22qter was re- Recent data indicate that some apparently quested. Chromosome studies suggested terminal chromosome deletions are, in fact, an abnormality involving the telomere of derivative chromosomes involving cryptic termi- one 22q (46,XX,?add(22)(q13.3)).