C/Ebpα Is an Essential Collaborator in Hoxa9/Meis1-Mediated Leukemogenesis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Homeobox Gene Expression Profile in Human Hematopoietic Multipotent
Leukemia (2003) 17, 1157–1163 & 2003 Nature Publishing Group All rights reserved 0887-6924/03 $25.00 www.nature.com/leu Homeobox gene expression profile in human hematopoietic multipotent stem cells and T-cell progenitors: implications for human T-cell development T Taghon1, K Thys1, M De Smedt1, F Weerkamp2, FJT Staal2, J Plum1 and G Leclercq1 1Department of Clinical Chemistry, Microbiology and Immunology, Ghent University Hospital, Ghent, Belgium; and 2Department of Immunology, Erasmus Medical Center, Rotterdam, The Netherlands Class I homeobox (HOX) genes comprise a large family of implicated in this transformation proces.14 The HOX-C locus transcription factors that have been implicated in normal and has been primarily implicated in lymphomas.15 malignant hematopoiesis. However, data on their expression or function during T-cell development is limited. Using degener- Hematopoietic cells are derived from stem cells that reside in ated RT-PCR and Affymetrix microarray analysis, we analyzed fetal liver (FL) in the embryo and in the adult bone marrow the expression pattern of this gene family in human multipotent (ABM), which have the unique ability to self-renew and thereby stem cells from fetal liver (FL) and adult bone marrow (ABM), provide a life-long supply of blood cells. T lymphocytes are a and in T-cell progenitors from child thymus. We show that FL specific type of hematopoietic cells that play a major role in the and ABM stem cells are similar in terms of HOX gene immune system. They develop through a well-defined order of expression, but significant differences were observed between differentiation steps in the thymus.16 Several transcription these two cell types and child thymocytes. -

Supplemental Materials ZNF281 Enhances Cardiac Reprogramming
Supplemental Materials ZNF281 enhances cardiac reprogramming by modulating cardiac and inflammatory gene expression Huanyu Zhou, Maria Gabriela Morales, Hisayuki Hashimoto, Matthew E. Dickson, Kunhua Song, Wenduo Ye, Min S. Kim, Hanspeter Niederstrasser, Zhaoning Wang, Beibei Chen, Bruce A. Posner, Rhonda Bassel-Duby and Eric N. Olson Supplemental Table 1; related to Figure 1. Supplemental Table 2; related to Figure 1. Supplemental Table 3; related to the “quantitative mRNA measurement” in Materials and Methods section. Supplemental Table 4; related to the “ChIP-seq, gene ontology and pathway analysis” and “RNA-seq” and gene ontology analysis” in Materials and Methods section. Supplemental Figure S1; related to Figure 1. Supplemental Figure S2; related to Figure 2. Supplemental Figure S3; related to Figure 3. Supplemental Figure S4; related to Figure 4. Supplemental Figure S5; related to Figure 6. Supplemental Table S1. Genes included in human retroviral ORF cDNA library. Gene Gene Gene Gene Gene Gene Gene Gene Symbol Symbol Symbol Symbol Symbol Symbol Symbol Symbol AATF BMP8A CEBPE CTNNB1 ESR2 GDF3 HOXA5 IL17D ADIPOQ BRPF1 CEBPG CUX1 ESRRA GDF6 HOXA6 IL17F ADNP BRPF3 CERS1 CX3CL1 ETS1 GIN1 HOXA7 IL18 AEBP1 BUD31 CERS2 CXCL10 ETS2 GLIS3 HOXB1 IL19 AFF4 C17ORF77 CERS4 CXCL11 ETV3 GMEB1 HOXB13 IL1A AHR C1QTNF4 CFL2 CXCL12 ETV7 GPBP1 HOXB5 IL1B AIMP1 C21ORF66 CHIA CXCL13 FAM3B GPER HOXB6 IL1F3 ALS2CR8 CBFA2T2 CIR1 CXCL14 FAM3D GPI HOXB7 IL1F5 ALX1 CBFA2T3 CITED1 CXCL16 FASLG GREM1 HOXB9 IL1F6 ARGFX CBFB CITED2 CXCL3 FBLN1 GREM2 HOXC4 IL1F7 -

1714 Gene Comprehensive Cancer Panel Enriched for Clinically Actionable Genes with Additional Biologically Relevant Genes 400-500X Average Coverage on Tumor
xO GENE PANEL 1714 gene comprehensive cancer panel enriched for clinically actionable genes with additional biologically relevant genes 400-500x average coverage on tumor Genes A-C Genes D-F Genes G-I Genes J-L AATK ATAD2B BTG1 CDH7 CREM DACH1 EPHA1 FES G6PC3 HGF IL18RAP JADE1 LMO1 ABCA1 ATF1 BTG2 CDK1 CRHR1 DACH2 EPHA2 FEV G6PD HIF1A IL1R1 JAK1 LMO2 ABCB1 ATM BTG3 CDK10 CRK DAXX EPHA3 FGF1 GAB1 HIF1AN IL1R2 JAK2 LMO7 ABCB11 ATR BTK CDK11A CRKL DBH EPHA4 FGF10 GAB2 HIST1H1E IL1RAP JAK3 LMTK2 ABCB4 ATRX BTRC CDK11B CRLF2 DCC EPHA5 FGF11 GABPA HIST1H3B IL20RA JARID2 LMTK3 ABCC1 AURKA BUB1 CDK12 CRTC1 DCUN1D1 EPHA6 FGF12 GALNT12 HIST1H4E IL20RB JAZF1 LPHN2 ABCC2 AURKB BUB1B CDK13 CRTC2 DCUN1D2 EPHA7 FGF13 GATA1 HLA-A IL21R JMJD1C LPHN3 ABCG1 AURKC BUB3 CDK14 CRTC3 DDB2 EPHA8 FGF14 GATA2 HLA-B IL22RA1 JMJD4 LPP ABCG2 AXIN1 C11orf30 CDK15 CSF1 DDIT3 EPHB1 FGF16 GATA3 HLF IL22RA2 JMJD6 LRP1B ABI1 AXIN2 CACNA1C CDK16 CSF1R DDR1 EPHB2 FGF17 GATA5 HLTF IL23R JMJD7 LRP5 ABL1 AXL CACNA1S CDK17 CSF2RA DDR2 EPHB3 FGF18 GATA6 HMGA1 IL2RA JMJD8 LRP6 ABL2 B2M CACNB2 CDK18 CSF2RB DDX3X EPHB4 FGF19 GDNF HMGA2 IL2RB JUN LRRK2 ACE BABAM1 CADM2 CDK19 CSF3R DDX5 EPHB6 FGF2 GFI1 HMGCR IL2RG JUNB LSM1 ACSL6 BACH1 CALR CDK2 CSK DDX6 EPOR FGF20 GFI1B HNF1A IL3 JUND LTK ACTA2 BACH2 CAMTA1 CDK20 CSNK1D DEK ERBB2 FGF21 GFRA4 HNF1B IL3RA JUP LYL1 ACTC1 BAG4 CAPRIN2 CDK3 CSNK1E DHFR ERBB3 FGF22 GGCX HNRNPA3 IL4R KAT2A LYN ACVR1 BAI3 CARD10 CDK4 CTCF DHH ERBB4 FGF23 GHR HOXA10 IL5RA KAT2B LZTR1 ACVR1B BAP1 CARD11 CDK5 CTCFL DIAPH1 ERCC1 FGF3 GID4 HOXA11 IL6R KAT5 ACVR2A -

Geminin Deletion Increases the Number of Fetal Hematopoietic Stem Cells by Affecting the Expression of Key Transcription Factors Dimitris Karamitros1,*, Alexandra L
© 2015. Published by The Company of Biologists Ltd | Development (2015) 142, 70-81 doi:10.1242/dev.109454 RESEARCH ARTICLE STEM CELLS AND REGENERATION Geminin deletion increases the number of fetal hematopoietic stem cells by affecting the expression of key transcription factors Dimitris Karamitros1,*, Alexandra L. Patmanidi1,*, Panoraia Kotantaki1, Alexandre J. Potocnik2, Tomi Bähr-Ivacevic3, Vladimir Benes3, Zoi Lygerou4, Dimitris Kioussis2,‡ and Stavros Taraviras1,‡ ABSTRACT hematological stress and challenges (Beerman et al., 2010; Balancing stem cell self-renewal and initiation of lineage specification Cheshier et al., 2007). The prevailing model of hematopoiesis programs is essential for the development and homeostasis of the supports the existence of long-term hematopoietic stem cells (LT- hematopoietic system. We have specifically ablated geminin in the HSCs), which can provide long-term multipotent reconstitution of developing murine hematopoietic system and observed profound the hematopoietic system, and of short-term hematopoietic stem defects in the generation of mature blood cells, leading to embryonic cells (ST-HSCs) or multipotent progenitors (MPPs), with the lethality. Hematopoietic stem cells (HSCs) accumulated in the fetal potential to generate all blood lineages but with reduced self- liver following geminin ablation, while committed progenitors were renewal capacity (Luc et al., 2007; Mebius et al., 2001; Morrison reduced. Genome-wide transcriptome analysis identified key HSC et al., 1995; Randall et al., 1996; Traver et al., 2001). transcription factors as being upregulated upon geminin deletion, Even though it remains unclear how the balance between self- revealing a gene network linked with geminin that controls fetal renewal of HSCs and fate commitment is controlled and how a hematopoiesis. -

Supplementary Materials
Supplementary Materials + - NUMB E2F2 PCBP2 CDKN1B MTOR AKT3 HOXA9 HNRNPA1 HNRNPA2B1 HNRNPA2B1 HNRNPK HNRNPA3 PCBP2 AICDA FLT3 SLAMF1 BIC CD34 TAL1 SPI1 GATA1 CD48 PIK3CG RUNX1 PIK3CD SLAMF1 CDKN2B CDKN2A CD34 RUNX1 E2F3 KMT2A RUNX1 T MIXL1 +++ +++ ++++ ++++ +++ 0 0 0 0 hematopoietic potential H1 H1 PB7 PB6 PB6 PB6.1 PB6.1 PB12.1 PB12.1 Figure S1. Unsupervised hierarchical clustering of hPSC-derived EBs according to the mRNA expression of hematopoietic lineage genes (microarray analysis). Hematopoietic-competent cells (H1, PB6.1, PB7) were separated from hematopoietic-deficient ones (PB6, PB12.1). In this experiment, all hPSCs were tested in duplicate, except PB7. Genes under-expressed or over-expressed in blood-deficient hPSCs are indicated in blue and red respectively (related to Table S1). 1 C) Mesoderm B) Endoderm + - KDR HAND1 GATA6 MEF2C DKK1 MSX1 GATA4 WNT3A GATA4 COL2A1 HNF1B ZFPM2 A) Ectoderm GATA4 GATA4 GSC GATA4 T ISL1 NCAM1 FOXH1 NCAM1 MESP1 CER1 WNT3A MIXL1 GATA4 PAX6 CDX2 T PAX6 SOX17 HBB NES GATA6 WT1 SOX1 FN1 ACTC1 ZIC1 FOXA2 MYF5 ZIC1 CXCR4 TBX5 PAX6 NCAM1 TBX20 PAX6 KRT18 DDX4 TUBB3 EPCAM TBX5 SOX2 KRT18 NKX2-5 NES AFP COL1A1 +++ +++ 0 0 0 0 ++++ +++ ++++ +++ +++ ++++ +++ ++++ 0 0 0 0 +++ +++ ++++ +++ ++++ 0 0 0 0 hematopoietic potential H1 H1 H1 H1 H1 H1 PB6 PB6 PB7 PB7 PB6 PB6 PB7 PB6 PB6 PB6.1 PB6.1 PB6.1 PB6.1 PB6.1 PB6.1 PB12.1 PB12.1 PB12.1 PB12.1 PB12.1 PB12.1 Figure S2. Unsupervised hierarchical clustering of hPSC-derived EBs according to the mRNA expression of germ layer differentiation genes (microarray analysis) Selected ectoderm (A), endoderm (B) and mesoderm (C) related genes differentially expressed between hematopoietic-competent (H1, PB6.1, PB7) and -deficient cells (PB6, PB12.1) are shown (related to Table S1). -

Test Summary Flyer-NGS Panels.Pub
Cancer‐related Mutaon Analysis Next Generaon Gene Sequencing for Myeloid Tesng Assay Summary IU Health Molecular Pathology Laboratory now offers high throughput sequencing for hot spot mutations found in clinically relevant cancer genes. In addition to a general panel of 54 genes, selected panels have been developed for a more tai- lored application in specific cancers. Comparing to single gene assay, these panels offer a more comprehensive and eco- nomic way to assess prognosis and/or treatment options for cancer patients at the initial diagnosis or at the relapse. Orderable Name: Use IU Health Molecular Pathology requisition; Call 317.491.6417 for requisition. Panels include: AML Mutations by NGS ASXL1, CEBPA, DNMT3A, ETV6/TEL, FLT3, HRAS, IDH1, IDH2, KIT, KRAS, MLL, NPM1, NRAS, PHF6, RUNX1, TET2, TP53, WT1 MDS Mutations by NGS ASXL1, ATRX, BCOR, BCORL1, ETV6/TEL, DNMT3A, EZH2, GNAS, IDH1, IDH2, RUNX1, SF3B1, SRSF2, TET2, TP53, U2AF1, ZRSR2 CML Mutations by NGS ABL1 MPN Mutations by NGS ASXL1, BRAF, CALR, CSF3R, EZH2, IKZF1, JAK2, JAK3, KDM6A, KIT, MPL, PDGRA, SETBP1, TET2 CMML Mutations by NGS ASXL1, CBL, CBLB, CBLC, EZH2, RUNX1, TET2, TP53, SRSF2 JMML Mutations by NGS CBL, CBLB, CBLC, HRAS, KRAS, NRAS, PTPN11 ALL Mutations by NGS ABL1, CSF3R, FBXW7, IKZF1, JAK3, KDM6A, NOTCH1 CLL Mutations by NGS MYD88, NOTCH1, SF3B1, TP53 Lymphoma/Myeloma Mutations by NGS BRAF, CDKN2A, CSF3R, FBXW7, HRAS, KRAS, MYD88, NOTCH1, NRAS, SF3B1,TP53 Hematopoietic Neoplasms Mutations by NGS ABL1, ASXL1, ATRX, BCOR, BCORL1, BRAF, CALR, CBL, CBLB, CBLC,CDKN2A, CEBPA, CSF3R, CUX1, DNMT3A, ETV6/TEL, EZH2, FBXW7, FLT3, GATA1, GATA2, GNAS, HRAS, IDH1, IDH2, IKZF1,JAK2, JAK3, KDM6A, KIT, KRAS, MLL, MPL, MYD88, NOTCH1, NPM1, NRAS, PDGFRA, PHF6, PTEN, PTPN11, RAD21, RUNX1,SETBP1, SF3B1, SMC1A, SMC3, SRSF2, STAG2, TET2, TP53, U2AF1, WT1, ZRSR2 Clinical Utility: This test is useful for the assessment of prognosis and/or treatment options for cancer patients at the initial diagnosis or at the relapse. -

HOXA10 Controls Osteoblastogenesis by Directly Activating Bone Regulatory and Phenotypic Genes
University of Massachusetts Medical School eScholarship@UMMS Open Access Articles Open Access Publications by UMMS Authors 2007-02-28 HOXA10 controls osteoblastogenesis by directly activating bone regulatory and phenotypic genes Mohammad Q. Hassan University of Massachusetts Medical School Et al. Let us know how access to this document benefits ou.y Follow this and additional works at: https://escholarship.umassmed.edu/oapubs Part of the Life Sciences Commons, and the Medicine and Health Sciences Commons Repository Citation Hassan MQ, Tare RS, Lee SH, Mandeville M, Weiner B, Montecino MA, Van Wijnen AJ, Stein JL, Stein GS, Lian JB. (2007). HOXA10 controls osteoblastogenesis by directly activating bone regulatory and phenotypic genes. Open Access Articles. https://doi.org/10.1128/MCB.01544-06. Retrieved from https://escholarship.umassmed.edu/oapubs/1334 This material is brought to you by eScholarship@UMMS. It has been accepted for inclusion in Open Access Articles by an authorized administrator of eScholarship@UMMS. For more information, please contact [email protected]. MOLECULAR AND CELLULAR BIOLOGY, May 2007, p. 3337–3352 Vol. 27, No. 9 0270-7306/07/$08.00ϩ0 doi:10.1128/MCB.01544-06 Copyright © 2007, American Society for Microbiology. All Rights Reserved. HOXA10 Controls Osteoblastogenesis by Directly Activating Bone Regulatory and Phenotypic Genesᰔ Mohammad Q. Hassan,1 Rahul Tare,1† Suk Hee Lee,1 Matthew Mandeville,1 Brian Weiner,1 Martin Montecino,2 Andre J. van Wijnen,1 Janet L. Stein,1 Gary S. Stein,1 and Jane B. Lian1* Department of Cell Biology and Cancer Center, University of Massachusetts Medical School, Worcester, Massachusetts 01655,1 and Departamento de Bioquimica y Biologia Molecular, Facultad de Ciencias Biologicas, Universidad de Concepcion, Concepcion, Chile2 Received 18 August 2006/Returned for modification 4 October 2006/Accepted 9 February 2007 HOXA10 is necessary for embryonic patterning of skeletal elements, but its function in bone formation beyond this early developmental stage is unknown. -
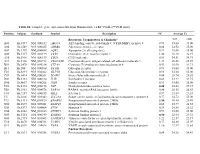
Table S1. Complete Gene Expression Data from Human Diabetes RT² Profiler™ PCR Array Receptors, Transporters & Channels* A
Table S1. Complete gene expression data from Human Diabetes RT² Profiler™ PCR Array Position Unigene GenBank Symbol Description FC Average Ct Receptors, Transporters & Channels* NGT GDM A01 Hs,5447 NM_000352 ABCC8 ATP-binding cassette, sub-family C (CFTR/MRP), member 8 0.93 35.00 35.00 A04 0Hs,2549 NM_000025 ADRB3 Adrenergic, beta-3-, receptor 0.88 34.92 35.00 A07 Hs,1307 NM_000486 AQP2 Aquaporin 2 (collecting duct) 0.93 35.00 35.00 A09 30Hs,5117 NM_001123 CCR2 Chemokine (C-C motif) receptor 2 1.00 26.28 26.17 A10 94Hs,5916 396NM_006139 CD28 CD28 molecule 0.81 34.51 34.71 A11 29Hs,5126 NM_001712 CEACAM1 Carcinoembryonic antigen-related cell adhesion molecule 1 1.31 26.08 25.59 B01 82Hs,2478 NM_005214 CTLA4 (biliaryCytotoxic glycoprotein) T-lymphocyte -associated protein 4 0.53 30.90 31.71 B11 24Hs,208 NM_000160 GCGR Glucagon receptor 0.93 35.00 35.00 C01 Hs,3891 NM_002062 GLP1R Glucagon-like peptide 1 receptor 0.93 35.00 35.00 C07 03Hs,6434 NM_000201 ICAM1 Intercellular adhesion molecule 1 0.84 28.74 28.89 D02 47Hs,5134 NM_000418 IL4R Interleukin 4 receptor 0.64 34.22 34.75 D06 57Hs,4657 NM_000208 INSR Insulin receptor 0.93 35.00 35.00 E05 44Hs,4312 NM_006178 NSF N-ethylmaleimide-sensitive factor 0.48 28.42 29.37 F08 79Hs,2961 NM_004578 RAB4A RAB4A, member RAS oncogene family 0.88 20.55 20.63 F10 69Hs,7287 NM_000655 SELL Selectin L 0.97 23.89 23.83 F11 56Hs,3806 NM_001042 SLC2A4 Solute carrier family 2 (facilitated glucose transporter), member 4 0.77 34.72 35.00 F12 91Hs,5111 NM_003825 SNAP23 Synaptosomal-associated protein, 23kDa 3.90 -

The Role of Constitutive Androstane Receptor and Estrogen Sulfotransferase in Energy Homeostasis
THE ROLE OF CONSTITUTIVE ANDROSTANE RECEPTOR AND ESTROGEN SULFOTRANSFERASE IN ENERGY HOMEOSTASIS by Jie Gao Bachelor of Engineering, China Pharmaceutical University, 1999 Master of Science, China Pharmaceutical University, 2002 Submitted to the Graduate Faculty of School of Pharmacy in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Pittsburgh 2012 UNIVERSITY OF PITTSBURGH School of Pharmacy This dissertation was presented by Jie Gao It was defended on January 18, 2012 and approved by Billy W. Day, Professor, Pharmaceutical Sciences Donald B. DeFranco, Professor, Pharmacology & Chemical Biology Samuel M. Poloyac, Associate Professor, Pharmaceutical Sciences Song Li, Associate Professor, Pharmaceutical Sciences Dissertation Advisor: Wen Xie, Professor, Pharmaceutical Sciences ii Copyright © by Jie Gao 2012 iii THE ROLE OF CONSTITUTIVE ANDROSTANE RECEPTOR AND ESTROGEN SULFOTRANSFERASE IN ENERGY HOMEOSTASIS Jie Gao, PhD University of Pittsburgh, 2012 Obesity and type 2 diabetes are related metabolic disorders of high prevalence. The constitutive androstane receptor (CAR) was initially characterized as a xenobiotic receptor regulating the responses of mammals to xenotoxicants. In this study, I have uncovered an unexpected role of CAR in preventing obesity and alleviating type 2 diabetes. Activation of CAR prevented obesity and improved insulin sensitivity in both the HFD-induced type 2 diabetic model and the ob/ob mice. In contrast, CAR null mice maintained on a chow diet showed spontaneous insulin insensitivity. The metabolic benefits of CAR activation may have resulted from inhibition of hepatic lipogenesis and gluconeogenesis. The molecular mechanism through which CAR activation suppressed hepatic gluconeogenesis might be mediated via peroxisome proliferator- activated receptor gamma coactivator-1 alpha (PGC-1α). -

Genequery™ Human Stem Cell Transcription Factors Qpcr Array
GeneQuery™ Human Stem Cell Transcription Factors qPCR Array Kit (GQH-SCT) Catalog #GK125 Product Description ScienCell's GeneQuery™ Human Stem Cell Transcription Factors qPCR Array Kit (GQH-SCT) surveys a panel of 88 transcription factors related to stem cell maintenance and differentiation. Transcription factors are proteins that can regulate target gene transcription level by binding to specific regions of the genome known as enhancers or silencers. Brief examples of how genes may be grouped according to their functions are shown below: • Pluripotency transcription factors: NANOG, POU5F1, SOX2 • Embryonic development: EOMES, FOXC2/D3/O1, HOXA9/A10, KLF2/5, LIN28B, MAX, PITX2, SMAD1, TBX5, ZIC1 • Germ layer formation and differentiation: FOXA2, GATA6, HAND1, ISL1, KLF4, SMAD2, SOX9 • Organ morphogenesis: EZH2, HOXA11/B3/B5/C9, MSX2, MYC, PAX5/8, VDR • Angiogenesis: CDX2, JUN, NR2F2, RUNX1, WT1 • Hematopoiesis and osteogenesis: EGR3, ESR1, GATA1/2/3, GLI2, NFATC1, PAX9, RB1, SOX6, SP1, STAT1 • Neural stem cell maintenance and differentiation: ATF2, CREB1, FOSB, FOXO3, HES1, HOXD10, MCM2/7, MEF2C, NEUROD1/G1, OLIG1/2, PAX3/6, POU4F1, PPARG, SMAD3/4, SNAI1, STAT3 Note : all gene names follow their official symbols by the Human Genome Organization Gene Nomenclature Committee (HGNC). GeneQuery™ qPCR array kits are qPCR ready in a 96-well plate format, with each well containing one primer set that can specifically recognize and efficiently amplify a target gene's cDNA. The carefully designed primers ensure that: (i) the optimal annealing temperature in qPCR analysis is 65°C (with 2 mM Mg 2+ , and no DMSO); (ii) the primer set recognizes all known transcript variants of target gene, unless otherwise indicated; and (iii) only one gene is amplified. -

Genetic Testing for Acute Myeloid Leukemia AHS-M2062
Corporate Medical Policy Genetic Testing for Acute Myeloid Leukemia AHS-M2062 File Name: genetic_testing_for_acute_myeloid_leukemia Origination: 1/1/2019 Last CAP Review: 8/2021 Next CAP Review: 8/2022 Last Review: 8/2021 Description of Procedure or Service Acute myeloid leukemia (AML) is characterized by large numbers of abnormal, immature myeloid cells in the bone marrow and peripheral blood resulting from genetic changes in hematopoietic precursor cells which disrupt normal hematopoietic growth and differentiation (Stock, 2020). Related Policies: Genetic Cancer Susceptibility Using Next Generation Sequencing AHS-M2066 Molecular Panel Testing of Cancers to Identify Targeted Therapy AHS-M2109 Serum Tumor Markers for Malignancies AHS-G2124 Minimal Residual Disease (MRD) AHS- M2175 ***Note: This Medical Policy is complex and technical. For questions concerning the technical language and/or specific clinical indications for its use, please consult your physician. Policy BCBSNC will provide coverage for genetic testing for acute myeloid leukemia when it is determined to be medically necessary because the medical criteria and guidelines shown below are met. Benefits Application This medical policy relates only to the services or supplies described herein. Please refer to the Member's Benefit Booklet for availability of benefits. Member's benefits may vary according to benefit design; therefore member benefit language should be reviewed before applying the terms of this medical policy. When Genetic Testing for Acute Myeloid Leukemia is covered The use of genetic testing for acute myeloid leukemia is considered medically necessary for the following: A. Genetic testing for FLT3 internal tandem duplication and tyrosine kinase domain mutations (ITD and TKD), IDH1, IDH2, TET2, WT1, DNMT3A, ASXL1 and/or TP53 in adult and pediatric patients with suspected or confirmed AML of any type for prognostic and/or therapeutic purposes. -

Epigenetic Therapy Restores Normal Hematopoiesis in a Zebrafish Model
Leukemia (2015) 29, 2086–2097 © 2015 Macmillan Publishers Limited All rights reserved 0887-6924/15 www.nature.com/leu ORIGINAL ARTICLE Epigenetic therapy restores normal hematopoiesis in a zebrafish model of NUP98–HOXA9-induced myeloid disease AP Deveau1,2,15, AM Forrester1,2,15, AJ Coombs2,3, GS Wagner2,3, C Grabher4,5, IC Chute6, D Léger6, M Mingay7, G Alexe4,8, V Rajan1,2, R Liwski9,10, M Hirst7,11, K Steigmaier4,8, SM Lewis1,6,12,13, AT Look4 and JN Berman1,2,10,14 Acute myeloid leukemia (AML) occurs when multiple genetic aberrations alter white blood cell development, leading to hyperproliferation and arrest of cell differentiation. Pertinent animal models link in vitro studies with the use of new agents in clinical trials. We generated a transgenic zebrafish expressing human NUP98–HOXA9 (NHA9), a fusion oncogene found in high-risk AML. Embryos developed a preleukemic state with anemia and myeloid cell expansion, and adult fish developed a myeloproliferative neoplasm (MPN). We leveraged this model to show that NHA9 increases the number of hematopoietic stem cells, and that oncogenic function of NHA9 depends on downstream activation of meis1, the PTGS/COX pathway and genome hypermethylation through the DNA methyltransferase, dnmt1. We restored normal hematopoiesis in NHA9 embryos with knockdown of meis1 or dnmt1, as well as pharmacologic treatment with DNA (cytosine-5)-methyltransferase (DNMT) inhibitors or cyclo-oxygenase (COX) inhibitors. DNMT inhibitors reduced genome methylation to near normal levels. Strikingly, we discovered synergy when we combined sub-monotherapeutic doses of a histone deacetylase inhibitor plus either a DNMT inhibitor or COX inhibitor to block the effects of NHA9 on zebrafish blood development.