Transcription Factor Assisted Loading and Enhancer Dynamics Dictate the Hepatic Fasting Response
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Down-Regulation of Stem Cell Genes, Including Those in a 200-Kb Gene Cluster at 12P13.31, Is Associated with in Vivo Differentiation of Human Male Germ Cell Tumors
Research Article Down-Regulation of Stem Cell Genes, Including Those in a 200-kb Gene Cluster at 12p13.31, Is Associated with In vivo Differentiation of Human Male Germ Cell Tumors James E. Korkola,1 Jane Houldsworth,1,2 Rajendrakumar S.V. Chadalavada,1 Adam B. Olshen,3 Debbie Dobrzynski,2 Victor E. Reuter,4 George J. Bosl,2 and R.S.K. Chaganti1,2 1Cell Biology Program and Departments of 2Medicine, 3Epidemiology and Biostatistics, and 4Pathology, Memorial Sloan-Kettering Cancer Center, New York, New York Abstract on the degree and type of differentiation (i.e., seminomas, which Adult male germ cell tumors (GCTs) comprise distinct groups: resemble undifferentiated primitive germ cells, and nonseminomas, seminomas and nonseminomas, which include pluripotent which show varying degrees of embryonic and extraembryonic embryonal carcinomas as well as other histologic subtypes patterns of differentiation; refs. 2, 3). Nonseminomatous GCTs are exhibiting various stages of differentiation. Almost all GCTs further subdivided into embryonal carcinomas, which show early show 12p gain, but the target genes have not been clearly zygotic or embryonal-like differentiation, yolk sac tumors and defined. To identify 12p target genes, we examined Affymetrix choriocarcinomas, which exhibit extraembryonal forms of differ- (Santa Clara, CA) U133A+B microarray (f83% coverage of 12p entiation, and teratomas, which show somatic differentiation along genes) expression profiles of 17 seminomas, 84 nonseminoma multiple lineages (3). Both seminomas and embryonal carcinoma GCTs, and 5 normal testis samples. Seventy-three genes on 12p are known to express stem cell markers, such as POU5F1 (4) and were significantly overexpressed, including GLUT3 and REA NANOG (5). -

Detection of Interacting Transcription Factors in Human Tissues Using
Myšičková and Vingron BMC Genomics 2012, 13(Suppl 1):S2 http://www.biomedcentral.com/1471-2164/13/S1/S2 PROCEEDINGS Open Access Detection of interacting transcription factors in human tissues using predicted DNA binding affinity Alena Myšičková*, Martin Vingron From The Tenth Asia Pacific Bioinformatics Conference (APBC 2012) Melbourne, Australia. 17-19 January 2012 Abstract Background: Tissue-specific gene expression is generally regulated by combinatorial interactions among transcription factors (TFs) which bind to the DNA. Despite this known fact, previous discoveries of the mechanism that controls gene expression usually consider only a single TF. Results: We provide a prediction of interacting TFs in 22 human tissues based on their DNA-binding affinity in promoter regions. We analyze all possible pairs of 130 vertebrate TFs from the JASPAR database. First, all human promoter regions are scanned for single TF-DNA binding affinities with TRAP and for each TF a ranked list of all promoters ordered by the binding affinity is created. We then study the similarity of the ranked lists and detect candidates for TF-TF interaction by applying a partial independence test for multiway contingency tables. Our candidates are validated by both known protein-protein interactions (PPIs) and known gene regulation mechanisms in the selected tissue. We find that the known PPIs are significantly enriched in the groups of our predicted TF-TF interactions (2 and 7 times more common than expected by chance). In addition, the predicted interacting TFs for studied tissues (liver, muscle, hematopoietic stem cell) are supported in literature to be active regulators or to be expressed in the corresponding tissue. -

Core Transcriptional Regulatory Circuitries in Cancer
Oncogene (2020) 39:6633–6646 https://doi.org/10.1038/s41388-020-01459-w REVIEW ARTICLE Core transcriptional regulatory circuitries in cancer 1 1,2,3 1 2 1,4,5 Ye Chen ● Liang Xu ● Ruby Yu-Tong Lin ● Markus Müschen ● H. Phillip Koeffler Received: 14 June 2020 / Revised: 30 August 2020 / Accepted: 4 September 2020 / Published online: 17 September 2020 © The Author(s) 2020. This article is published with open access Abstract Transcription factors (TFs) coordinate the on-and-off states of gene expression typically in a combinatorial fashion. Studies from embryonic stem cells and other cell types have revealed that a clique of self-regulated core TFs control cell identity and cell state. These core TFs form interconnected feed-forward transcriptional loops to establish and reinforce the cell-type- specific gene-expression program; the ensemble of core TFs and their regulatory loops constitutes core transcriptional regulatory circuitry (CRC). Here, we summarize recent progress in computational reconstitution and biologic exploration of CRCs across various human malignancies, and consolidate the strategy and methodology for CRC discovery. We also discuss the genetic basis and therapeutic vulnerability of CRC, and highlight new frontiers and future efforts for the study of CRC in cancer. Knowledge of CRC in cancer is fundamental to understanding cancer-specific transcriptional addiction, and should provide important insight to both pathobiology and therapeutics. 1234567890();,: 1234567890();,: Introduction genes. Till now, one critical goal in biology remains to understand the composition and hierarchy of transcriptional Transcriptional regulation is one of the fundamental mole- regulatory network in each specified cell type/lineage. -

Derivation of Stable Microarray Cancer-Differentiating Signatures Using Consensus Scoring of Multiple Random Sampling and Gene-Ranking Consistency Evaluation
Research Article Derivation of Stable Microarray Cancer-Differentiating Signatures Using Consensus Scoring of Multiple Random Sampling and Gene-Ranking Consistency Evaluation Zhi Qun Tang,1,2 Lian Yi Han,1,2 Hong Huang Lin,1,2 Juan Cui,1,2 Jia Jia,1,2 Boon Chuan Low,2,3 Bao Wen Li,2,4 and Yu Zong Chen1,2 1Bioinformatics and Drug Design Group, Department of Pharmacy; 2Center for Computational Science and Engineering; and Departments of 3Biological Sciences and 4Physics, National University of Singapore, Singapore, Singapore Abstract sampling methods. Only 1 to 5 of the 4 to 60 selected predictor Microarrays have been explored for deriving molecular genes in each of these sets are present in more than half of the signatures to determine disease outcomes, mechanisms, other nine sets (Table 1), and 2 to 20 of the predictor genes in each targets, and treatment strategies. Although exhibiting good set are cancer related (Table 2). Despite the use of sophisticated predictive performance, some derived signatures are unstable class differentiation and signature selection methods, the selected due to noises arising from measurement variability and signatures show few overlapping predictor genes, as in the case of biological differences. Improvements in measurement, anno- other microarray data sets including non–Hodgkin lymphoma, tation, and signature selection methods have been proposed. acute lymphocytic leukemia, breast cancer, lung adenocarcinoma, We explored a new signature selection method that incorpo- medulloblastoma, hepatocellular carcinoma, and acute myeloid rates consensus scoring of multiple random sampling and leukemia (9, 15). multistep evaluation of gene-ranking consistency for maxi- Although these signatures display high cancer differentiation mally avoiding erroneous elimination of predictor genes. -
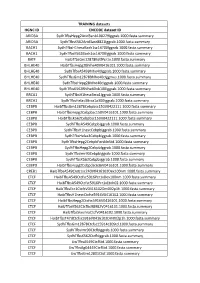
TRAINING Datasets HGNC ID ENCODE Dataset ID ARID3A
TRAINING datasets HGNC ID ENCODE dataset ID ARID3A SydhT+sHepg2Arid3anb100279Iggrab.1000.fasta.summary ARID3A SydhT+sK562Arid3asC8821Iggrab.1000.fasta.summary BACH1 SydhT+sH1hesCBaCh1sC14700Iggrab.1000.fasta.summary BACH1 SydhT+sK562BaCh1sC14700Iggrab.1000.fasta.summary BATF HaibT+sGm12878BaJPCr1x.1000.fasta.summary BHLHE40 HaibT+sHepg2Bhlhe40V0416101.1000.fasta.summary BHLHE40 SydhT+sA549Bhlhe40Iggrab.1000.fasta.summary BHLHE40 SydhT+sGm12878Bhlhe40CIggmus.1000.fasta.summary BHLHE40 SydhT+sHepg2Bhlhe40CIggrab.1000.fasta.summary BHLHE40 SydhT+sK562Bhlhe40nb100Iggrab.1000.fasta.summary BRCA1 SydhT+sH1hesCBrCa1Iggrab.1000.fasta.summary BRCA1 SydhT+sHelas3BrCa1a300Iggrab.1000.fasta.summary CEBPB HaibT+sGm12878CebpbsC150V0422111.1000.fasta.summary CEBPB HaibT+sHepg2CebpbsC150V0416101.1000.fasta.summary CEBPB HaibT+sK562CebpbsC150V0422111.1000.fasta.summary CEBPB SydhT+sA549CebpbIggrab.1000.fasta.summary CEBPB SydhT+sH1hesCCebpbIggrab.1000.fasta.summary CEBPB SydhT+sHelas3CebpbIggrab.1000.fasta.summary CEBPB SydhT+sHepg2CebpbForsklnStd.1000.fasta.summary CEBPB SydhT+sHepg2CebpbIggrab.1000.fasta.summary CEBPB SydhT+sImr90CebpbIggrab.1000.fasta.summary CEBPB SydhT+sK562CebpbIggrab.1000.fasta.summary CEBPD HaibT+sHepg2CebpdsC636V0416101.1000.fasta.summary CREB1 HaibT+sA549Creb1sC240V0416102Dex100nm.1000.fasta.summary CTCF HaibT+sA549CtCfsC5916PCr1xDex100nm.1000.fasta.summary CTCF HaibT+sA549CtCfsC5916PCr1xEtoh02.1000.fasta.summary CTCF HaibT+sECC1CtCfCV0416102Dm002p1h.1000.fasta.summary CTCF HaibT+sH1hesCCtCfsC5916V0416102.1000.fasta.summary -

Jmjd2c Facilitates the Assembly of Essential Enhancer-Protein Complexes at the Onset of Embryonic Stem Cell Differentiation Rute A
© 2017. Published by The Company of Biologists Ltd | Development (2017) 144, 567-579 doi:10.1242/dev.142489 STEM CELLS AND REGENERATION RESEARCH ARTICLE Jmjd2c facilitates the assembly of essential enhancer-protein complexes at the onset of embryonic stem cell differentiation Rute A. Tomaz1,JenniferL.Harman2, Donja Karimlou1, Lauren Weavers1, Lauriane Fritsch3, Tony Bou-Kheir1, Emma Bell1, Ignacio del Valle Torres4, Kathy K. Niakan4,CynthiaFisher5, Onkar Joshi6, Hendrik G. Stunnenberg6, Edward Curry1, Slimane Ait-Si-Ali3, Helle F. Jørgensen2 and Véronique Azuara1,* ABSTRACT implantation, a second extra-embryonic lineage, the primitive Jmjd2 H3K9 demethylases cooperate in promoting mouse embryonic endoderm, emerges at the ICM surface. Concurrently, the ICM stem cell (ESC) identity. However, little is known about their maintains its pluripotency as it matures into the epiblast but importance at the exit of ESC pluripotency. Here, we reveal that ultimately goes on to form the three primary germ layers and germ Jmjd2c facilitates this process by stabilising the assembly of cells upon gastrulation (Boroviak and Nichols, 2014; Rossant, mediator-cohesin complexes at lineage-specific enhancers. 2008). Functionally, we show that Jmjd2c is required in ESCs to initiate Pluripotent mouse embryonic stem cells (ESCs) are derived from appropriate gene expression programs upon somatic multi-lineage ICM cells, and can self-renew and faithfully maintain an differentiation. In the absence of Jmjd2c, differentiation is stalled at an undifferentiated state in vitro in the presence of leukaemia inhibitory early post-implantation epiblast-like stage, while Jmjd2c-knockout factor (LIF) and serum components, while preserving their multi- ESCs remain capable of forming extra-embryonic endoderm lineage differentiation capacity (Evans and Kaufman, 1981; Martin, derivatives. -

2017.08.28 Anne Barry-Reidy Thesis Final.Pdf
REGULATION OF BOVINE β-DEFENSIN EXPRESSION THIS THESIS IS SUBMITTED TO THE UNIVERSITY OF DUBLIN FOR THE DEGREE OF DOCTOR OF PHILOSOPHY 2017 ANNE BARRY-REIDY SCHOOL OF BIOCHEMISTRY & IMMUNOLOGY TRINITY COLLEGE DUBLIN SUPERVISORS: PROF. CLIONA O’FARRELLY & DR. KIERAN MEADE TABLE OF CONTENTS DECLARATION ................................................................................................................................. vii ACKNOWLEDGEMENTS ................................................................................................................... viii ABBREVIATIONS ................................................................................................................................ix LIST OF FIGURES............................................................................................................................. xiii LIST OF TABLES .............................................................................................................................. xvii ABSTRACT ........................................................................................................................................xix Chapter 1 Introduction ........................................................................................................ 1 1.1 Antimicrobial/Host-defence peptides ..................................................................... 1 1.2 Defensins................................................................................................................. 1 1.3 β-defensins ............................................................................................................. -

Early B-Cell Factors Are Required for Specifying Multiple Retinal Cell Types and Subtypes from Postmitotic Precursors
11902 • The Journal of Neuroscience, September 8, 2010 • 30(36):11902–11916 Development/Plasticity/Repair Early B-Cell Factors Are Required for Specifying Multiple Retinal Cell Types and Subtypes from Postmitotic Precursors Kangxin Jin,1,2 Haisong Jiang,1,2 Zeqian Mo,3 and Mengqing Xiang1,2 1Center for Advanced Biotechnology and Medicine and Department of Pediatrics, 2Graduate Program in Molecular Genetics, Microbiology and Immunology, and 3Department of Cell Biology and Neuroscience, University of Medicine and Dentistry of New Jersey-Robert Wood Johnson Medical School, Piscataway, New Jersey 08854 The establishment of functional retinal circuits in the mammalian retina depends critically on the proper generation and assembly of six classes of neurons, five of which consist of two or more subtypes that differ in morphologies, physiological properties, and/or sublaminar positions. How these diverse neuronal types and subtypes arise during retinogenesis still remains largely to be defined at the molecular level. Here we show that all four family members of the early B-cell factor (Ebf) helix-loop-helix transcription factors are similarly expressedduringmouseretinogenesisinseveralneuronaltypesandsubtypesincludingganglion,amacrine,bipolar,andhorizontalcells, and that their expression in ganglion cells depends on the ganglion cell specification factor Brn3b. Misexpressed Ebfs bias retinal precursors toward the fates of non-AII glycinergic amacrine, type 2 OFF-cone bipolar and horizontal cells, whereas a dominant-negative Ebf suppresses the differentiation of these cells as well as ganglion cells. Reducing Ebf1 expression by RNA interference (RNAi) leads to an inhibitory effect similar to that of the dominant-negative Ebf, effectively neutralizes the promotive effect of wild-type Ebf1, but has no impact on the promotive effect of an RNAi-resistant Ebf1. -

Tolerance to Sustained Activation of the Camp/Creb Pathway Activity in Osteoblastic Cells Is Enabled by Loss of P53 Mannu K
Walia et al. Cell Death and Disease (2018) 9:844 DOI 10.1038/s41419-018-0944-8 Cell Death & Disease ARTICLE Open Access Tolerance to sustained activation of the cAMP/Creb pathway activity in osteoblastic cells is enabled by loss of p53 Mannu K. Walia1, Scott Taylor1,PatriciaW.M.Ho1,T.JohnMartin1,2 and Carl R. Walkley 1,2,3 Abstract The loss of p53 function is a central event in the genesis of osteosarcoma (OS). How mutation of p53 enables OS development from osteoblastic lineage cells is poorly understood. We and others have reported a key role for elevated and persistent activation of the cAMP/PKA/Creb1 pathway in maintenance of OS. In view of the osteoblast lineage being the cell of origin of OS, we sought to determine how these pathways interact within the context of the normal osteoblast. Normal osteoblasts (p53 WT) rapidly underwent apoptosis in response to acute elevation of cAMP levels or activity, whereas p53-deficient osteoblasts tolerated this aberrant cAMP/Creb level and activity. Using the p53 activating small-molecule Nutlin-3a and cAMP/Creb1 activator forskolin, we addressed the question of how p53 responds to the activation of cAMP. We observed that p53 acts dominantly to protect cells from excessive cAMP accumulation. We identify a Creb1-Cbp complex that functions together with and interacts with p53. Finally, translating these results we find that a selective small-molecule inhibitor of the Creb1-Cbp interaction demonstrates selective toxicity to OS cells where this pathway is constitutively active. This highlights the cAMP/Creb axis as a potentially actionable therapeutic vulnerability in p53-deficient tumors such as OS. -

Farnesol-Induced Apoptosis in Human Lung Carcinoma Cells Is Coupled to the Endoplasmic Reticulum Stress Response
Research Article Farnesol-Induced Apoptosis in Human Lung Carcinoma Cells Is Coupled to the Endoplasmic Reticulum Stress Response Joung Hyuck Joo,1 Grace Liao,1 Jennifer B. Collins,2 Sherry F. Grissom,2 and Anton M. Jetten1 1Cell Biology Section, LRB, and 2Microarray Group, Division of Intramural Research, National Institute of Environmental Health Sciences, NIH, Research Triangle Park, North Carolina Abstract range of fruits and vegetables (9, 10). Each isoprenoid has been Farnesol (FOH) and other isoprenoid alcohols induce apopto- shown to inhibit proliferation and induce apoptosis in a number of sis in various carcinoma cells and inhibit tumorigenesis in neoplastic cell lines from different origins (4, 11–14). In addition, in vivo these isoprenoids have been reported to be effective in chemo- several models. However, the mechanisms by which in vivo they mediate their effects are not yet fully understood. In this prevention and chemotherapy in various cancer models study, we show that FOH is an effective inducer of apoptosis in (10, 12, 15, 16). FOH has been reported to exhibit chemopreventive several lung carcinoma cells, including H460. This induction is effects in colon and pancreas carcinogenesis in rats (9, 17) whereas associated with activation of several caspases and cleavage of phase I and II clinical trials have indicated therapeutic potential poly(ADP-ribose) polymerase (PARP). To obtain insight into for POH (16, 18). The mechanisms by which these isoprenoids induce these effects are not yet fully understood. Isoprenoids have the mechanism involved in FOH-induced apoptosis, we compared the gene expression profiles of FOH-treated and been reported to inhibit posttranslational protein prenylation (19) control H460 cells by microarray analysis. -

Identification of BBX Proteins As Rate-Limiting Co-Factors of HY5
1 Identification of BBX proteins as rate-limiting co-factors of HY5. 2 Katharina Bursch1, Gabriela Toledo-Ortiz2, Marie Pireyre3, Miriam Lohr1, Cordula Braatz1, Henrik 3 Johansson1* 4 1. Institute of Biology/Applied Genetics, Dahlem Centre of Plant Sciences (DCPS), Freie Universität 5 Berlin, Albrecht-Thaer-Weg 6. D-14195 Berlin, Germany. 6 2. Lancaster Environment Centre, Lancaster University, Lancaster LA1 4YQ, UK 7 3. Department of Botany and Plant Biology, Section of Biology, Faculty of Sciences, University of 8 Geneva, 30 Quai E. Ansermet, 1211 Geneva 4, Switzerland 9 *E-mail [email protected] 10 Abstract 11 As a source of both energy and environmental information, monitoring the incoming light is crucial for 12 plants to optimize growth throughout development1. Concordantly, the light signalling pathways in 13 plants are highly integrated with numerous other regulatory pathways2,3. One of these signal 14 integrators is the bZIP transcription factor HY5 which holds a key role as a positive regulator of light 15 signalling in plants4,5. Although HY5 is thought to act as a DNA-binding transcriptional regulator6,7, the 16 lack of any apparent transactivation domain8 makes it unclear how HY5 is able to accomplish its many 17 functions. Here, we describe the identification of three B-box containing proteins (BBX20, 21 and 22) 18 as essential partners for HY5 dependent modulation of hypocotyl elongation, anthocyanin 19 accumulation and transcriptional regulation. The bbx202122 triple mutant mimics the phenotypes of 20 hy5 in the light and its ability to suppress the cop1 mutant phenotype in darkness. Furthermore, 84% 21 of genes that exhibit differential expression in bbx202122 are also HY5 regulated, and we provide 22 evidence that HY5 requires the B-box proteins for transcriptional regulation. -

Supplemental Materials ZNF281 Enhances Cardiac Reprogramming
Supplemental Materials ZNF281 enhances cardiac reprogramming by modulating cardiac and inflammatory gene expression Huanyu Zhou, Maria Gabriela Morales, Hisayuki Hashimoto, Matthew E. Dickson, Kunhua Song, Wenduo Ye, Min S. Kim, Hanspeter Niederstrasser, Zhaoning Wang, Beibei Chen, Bruce A. Posner, Rhonda Bassel-Duby and Eric N. Olson Supplemental Table 1; related to Figure 1. Supplemental Table 2; related to Figure 1. Supplemental Table 3; related to the “quantitative mRNA measurement” in Materials and Methods section. Supplemental Table 4; related to the “ChIP-seq, gene ontology and pathway analysis” and “RNA-seq” and gene ontology analysis” in Materials and Methods section. Supplemental Figure S1; related to Figure 1. Supplemental Figure S2; related to Figure 2. Supplemental Figure S3; related to Figure 3. Supplemental Figure S4; related to Figure 4. Supplemental Figure S5; related to Figure 6. Supplemental Table S1. Genes included in human retroviral ORF cDNA library. Gene Gene Gene Gene Gene Gene Gene Gene Symbol Symbol Symbol Symbol Symbol Symbol Symbol Symbol AATF BMP8A CEBPE CTNNB1 ESR2 GDF3 HOXA5 IL17D ADIPOQ BRPF1 CEBPG CUX1 ESRRA GDF6 HOXA6 IL17F ADNP BRPF3 CERS1 CX3CL1 ETS1 GIN1 HOXA7 IL18 AEBP1 BUD31 CERS2 CXCL10 ETS2 GLIS3 HOXB1 IL19 AFF4 C17ORF77 CERS4 CXCL11 ETV3 GMEB1 HOXB13 IL1A AHR C1QTNF4 CFL2 CXCL12 ETV7 GPBP1 HOXB5 IL1B AIMP1 C21ORF66 CHIA CXCL13 FAM3B GPER HOXB6 IL1F3 ALS2CR8 CBFA2T2 CIR1 CXCL14 FAM3D GPI HOXB7 IL1F5 ALX1 CBFA2T3 CITED1 CXCL16 FASLG GREM1 HOXB9 IL1F6 ARGFX CBFB CITED2 CXCL3 FBLN1 GREM2 HOXC4 IL1F7