Monitoring of Residual Disease by Next-Generation Deep-Sequencing of RUNX1 Mutations Can Identify Acute Myeloid Leukemia Patients with Resistant Disease
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Minimal Residual Disease Detection in Childhood Precursor–B-Cell Acute Lymphoblastic Leukemia: Relation to Other Risk Factors
Leukemia (2003) 17, 1566–1572 & 2003 Nature Publishing Group All rights reserved 0887-6924/03 $25.00 www.nature.com/leu Minimal residual disease detection in childhood precursor–B-cell acute lymphoblastic leukemia: relation to other risk factors. A Children’s Oncology Group study MJ Borowitz1, DJ Pullen2, JJ Shuster3,4, D Viswanatha5, K Montgomery6, CL Willman5 and B Camitta7 1Johns Hopkins Medical Institutions, USA; 2University of Mississippi, USA; 3University of Florida, Gainesville FL, USA; 4Children’s Oncology Group, Arcadia, CA, USA; 5University of New Mexico, USA; 6Genzyme Genetics, Santa Fe, NM; and 7Milwaukee Childrens Hospital, USA Minimal residual disease (MRD) can be detected in the marrows prognostic significance at several time points. MRD levels at the of children undergoing chemotherapy either by flow cytometry end of induction are highly prognostic. Since intensification of or polymerase chain reaction. In this study, we used four-color therapy after successful induction can improve the outcome of flow cytometry to detect MRD in 1016 children undergoing 33 therapy on Children’s Oncology Group therapeutic protocols many patients with adverse prognostic features, it is possible for precursor–B-cell ALL. Compliance was excellent, with that a similar approach might benefit patients with MRD. For follow-up samples received at the end of induction on nearly these reasons, we have chosen to focus our MRD studies on 95% of cases; sensitivity of detection at this time point was at end-of-induction therapy. least 1/10,000 in more than 90% of cases. Overall, 28.6% of Although MRD and other measures of early response to patients had detectable MRD at the end of induction. -

C/Ebpα Is an Essential Collaborator in Hoxa9/Meis1-Mediated Leukemogenesis
C/EBPα is an essential collaborator in Hoxa9/Meis1-mediated leukemogenesis Cailin Collinsa, Jingya Wanga, Hongzhi Miaoa, Joel Bronsteina, Humaira Nawera, Tao Xua, Maria Figueroaa, Andrew G. Munteana, and Jay L. Hessa,b,1 aDepartment of Pathology, University of Michigan, Ann Arbor, MI 48109; and bIndiana University School of Medicine, Indianapolis, IN 46202 Edited* by Louis M. Staudt, National Institutes of Health, Bethesda, MD, and approved May 19, 2014 (received for review February 12, 2014) Homeobox A9 (HOXA9) is a homeodomain-containing transcrip- with Hoxa9. In addition, C/EBP recognition motifs are enriched tion factor that plays a key role in hematopoietic stem cell expan- at Hoxa9 binding sites. sion and is commonly deregulated in human acute leukemias. A C/EBPα is a basic leucine-zipper transcription factor that plays variety of upstream genetic alterations in acute myeloid leukemia a critical role in lineage commitment during hematopoietic dif- −/− (AML) lead to overexpression of HOXA9, almost always in associ- ferentiation (18). Whereas Cebpa mice show complete loss of ation with overexpression of its cofactor meis homeobox 1 (MEIS1). the granulocytic compartment, recent work shows that loss of α A wide range of data suggests that HOXA9 and MEIS1 play a syn- C/EBP in adult HSCs leads to both an increase in the number ergistic causative role in AML, although the molecular mechanisms of functional HSCs and an increase in their proliferative and leading to transformation by HOXA9 and MEIS1 remain elusive. In repopulating capacity (19, 20). Conversely, CEBPA overexpression can promote transdifferentiation of a variety of fibroblastic cells to this study, we identify CCAAT/enhancer binding protein alpha (C/ the myeloid lineage and can induce monocytic differentiation in EBPα) as a critical collaborator required for Hoxa9/Meis1-mediated α MLL-fusion protein-mediated leukemias (21, 22). -

Effect of the Philadelphia Chromosome on Minimal Residual Disease In
Leukemia (1997) 11, 1497–1500 1997 Stockton Press All rights reserved 0887-6924/97$12.00 Effect of the Philadelphia chromosome on minimal residual disease in acute lymphoblastic leukemia MJ Brisco1, PJ Sykes1, G Dolman1, S-H Neoh1, E Hughes1, L-M Peng2, G Tauro3, H Ekert3, I Toogood4, K Bradstock5 and AA Morley1 1Department of Haematology, Flinders Medical Centre, Bedford Park, South Australia; 2Department of Laboratory Medicine, School of Medicine, West China University of Medical Sciences, Chengdu, People’s Republic of China; 3Department of Haematology, Royal Childrens Hospital, Parkville, Victoria; 4Department of Haematology/Oncology, Womens and Childrens Hospital, North Adelaide, South Australia; and 5Department of Haematology, Westmead Hospital, Westmead, NSW, Australia The Philadelphia translocation is associated with a poor prog- number of leukemic cells remaining at the end of induction nosis in adults and children with acute lymphoblastic leukemia, provides a good approximation of the degree of drug resist- even though the majority of patients achieve remission. To test ance in vivo. the hypothesis that the translocation leads to drug resistance 6–10 in vivo, we studied 61 children and 20 adults with acute lym- Since the initial reports, a number of groups have phoblastic leukemia and used the level of minimal residual dis- developed sensitive methods to quantify minimal residual dis- ease at the end of induction as the measure of drug resistance ease (MRD) by use of the polymerase chain reaction (PCR). in vivo. In children -

Test Summary Flyer-NGS Panels.Pub
Cancer‐related Mutaon Analysis Next Generaon Gene Sequencing for Myeloid Tesng Assay Summary IU Health Molecular Pathology Laboratory now offers high throughput sequencing for hot spot mutations found in clinically relevant cancer genes. In addition to a general panel of 54 genes, selected panels have been developed for a more tai- lored application in specific cancers. Comparing to single gene assay, these panels offer a more comprehensive and eco- nomic way to assess prognosis and/or treatment options for cancer patients at the initial diagnosis or at the relapse. Orderable Name: Use IU Health Molecular Pathology requisition; Call 317.491.6417 for requisition. Panels include: AML Mutations by NGS ASXL1, CEBPA, DNMT3A, ETV6/TEL, FLT3, HRAS, IDH1, IDH2, KIT, KRAS, MLL, NPM1, NRAS, PHF6, RUNX1, TET2, TP53, WT1 MDS Mutations by NGS ASXL1, ATRX, BCOR, BCORL1, ETV6/TEL, DNMT3A, EZH2, GNAS, IDH1, IDH2, RUNX1, SF3B1, SRSF2, TET2, TP53, U2AF1, ZRSR2 CML Mutations by NGS ABL1 MPN Mutations by NGS ASXL1, BRAF, CALR, CSF3R, EZH2, IKZF1, JAK2, JAK3, KDM6A, KIT, MPL, PDGRA, SETBP1, TET2 CMML Mutations by NGS ASXL1, CBL, CBLB, CBLC, EZH2, RUNX1, TET2, TP53, SRSF2 JMML Mutations by NGS CBL, CBLB, CBLC, HRAS, KRAS, NRAS, PTPN11 ALL Mutations by NGS ABL1, CSF3R, FBXW7, IKZF1, JAK3, KDM6A, NOTCH1 CLL Mutations by NGS MYD88, NOTCH1, SF3B1, TP53 Lymphoma/Myeloma Mutations by NGS BRAF, CDKN2A, CSF3R, FBXW7, HRAS, KRAS, MYD88, NOTCH1, NRAS, SF3B1,TP53 Hematopoietic Neoplasms Mutations by NGS ABL1, ASXL1, ATRX, BCOR, BCORL1, BRAF, CALR, CBL, CBLB, CBLC,CDKN2A, CEBPA, CSF3R, CUX1, DNMT3A, ETV6/TEL, EZH2, FBXW7, FLT3, GATA1, GATA2, GNAS, HRAS, IDH1, IDH2, IKZF1,JAK2, JAK3, KDM6A, KIT, KRAS, MLL, MPL, MYD88, NOTCH1, NPM1, NRAS, PDGFRA, PHF6, PTEN, PTPN11, RAD21, RUNX1,SETBP1, SF3B1, SMC1A, SMC3, SRSF2, STAG2, TET2, TP53, U2AF1, WT1, ZRSR2 Clinical Utility: This test is useful for the assessment of prognosis and/or treatment options for cancer patients at the initial diagnosis or at the relapse. -

REVIEW Clinical Relevance of Minimal Residual Disease Monitoring in Non
Leukemia (1999) 13, 1691–1695 1999 Stockton Press All rights reserved 0887-6924/99 $15.00 http://www.stockton-press.co.uk/leu REVIEW Clinical relevance of minimal residual disease monitoring in non-Hodgkin’s lymphomas: a critical reappraisal of molecular strategies P Corradini1, M Ladetto2, A Pileri2 and C Tarella2 1Bone Marrow Transplantation Unit, Istituto Scientifico HS Raffaele, Milan; and 2Divisione Universitaria di Ematologia–Azienda Ospedaliera S Giovanni Battista, Turin, Italy Although current treatments can induce clinical complete neoplasms. In the NHL setting, several studies have been pub- remissions in the vast majority of patients with indolent lym- lished on the prognostic significance of minimal residual dis- phoma, most of them actually relapse, because of the persist- ence of residual tumor cells which are undetectable using con- ease (MRD) detection. Most of these studies focus on indolent ventional diagnostic procedures. Polymerase chain reaction lymphomas: small lymphocytic lymphoma/chronic lympho- (PCR)-based methods are increasingly used for minimal cytic leukemia (SLL/CLL),17,18 follicular (FCL)19–21 and mantle residual disease detection (MRD), and provide useful prognos- cell lymphomas (MCL).20,22 This is mostly because these tic information. In this review, current approaches for MRD tumors, unlike more aggressive NHL histotypes, are dissemi- detection in indolent lymphomas are summarized. In addition, nated disorders with frequent microscopic or submicroscopic the prognostic aspects of molecular monitoring after transplan- tation procedures are discussed. The experience accumulated bone marrow (BM) and peripheral blood (PB) involvement, over the past decade shows that PCR analysis has a prognostic and thus they represent ideal targets for MRD evaluation with impact in several therapeutic programs including conventional PCR-based assays.23,24 and high-dose regimens. -
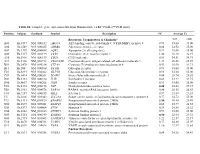
Table S1. Complete Gene Expression Data from Human Diabetes RT² Profiler™ PCR Array Receptors, Transporters & Channels* A
Table S1. Complete gene expression data from Human Diabetes RT² Profiler™ PCR Array Position Unigene GenBank Symbol Description FC Average Ct Receptors, Transporters & Channels* NGT GDM A01 Hs,5447 NM_000352 ABCC8 ATP-binding cassette, sub-family C (CFTR/MRP), member 8 0.93 35.00 35.00 A04 0Hs,2549 NM_000025 ADRB3 Adrenergic, beta-3-, receptor 0.88 34.92 35.00 A07 Hs,1307 NM_000486 AQP2 Aquaporin 2 (collecting duct) 0.93 35.00 35.00 A09 30Hs,5117 NM_001123 CCR2 Chemokine (C-C motif) receptor 2 1.00 26.28 26.17 A10 94Hs,5916 396NM_006139 CD28 CD28 molecule 0.81 34.51 34.71 A11 29Hs,5126 NM_001712 CEACAM1 Carcinoembryonic antigen-related cell adhesion molecule 1 1.31 26.08 25.59 B01 82Hs,2478 NM_005214 CTLA4 (biliaryCytotoxic glycoprotein) T-lymphocyte -associated protein 4 0.53 30.90 31.71 B11 24Hs,208 NM_000160 GCGR Glucagon receptor 0.93 35.00 35.00 C01 Hs,3891 NM_002062 GLP1R Glucagon-like peptide 1 receptor 0.93 35.00 35.00 C07 03Hs,6434 NM_000201 ICAM1 Intercellular adhesion molecule 1 0.84 28.74 28.89 D02 47Hs,5134 NM_000418 IL4R Interleukin 4 receptor 0.64 34.22 34.75 D06 57Hs,4657 NM_000208 INSR Insulin receptor 0.93 35.00 35.00 E05 44Hs,4312 NM_006178 NSF N-ethylmaleimide-sensitive factor 0.48 28.42 29.37 F08 79Hs,2961 NM_004578 RAB4A RAB4A, member RAS oncogene family 0.88 20.55 20.63 F10 69Hs,7287 NM_000655 SELL Selectin L 0.97 23.89 23.83 F11 56Hs,3806 NM_001042 SLC2A4 Solute carrier family 2 (facilitated glucose transporter), member 4 0.77 34.72 35.00 F12 91Hs,5111 NM_003825 SNAP23 Synaptosomal-associated protein, 23kDa 3.90 -

Genetic Testing for Acute Myeloid Leukemia AHS-M2062
Corporate Medical Policy Genetic Testing for Acute Myeloid Leukemia AHS-M2062 File Name: genetic_testing_for_acute_myeloid_leukemia Origination: 1/1/2019 Last CAP Review: 8/2021 Next CAP Review: 8/2022 Last Review: 8/2021 Description of Procedure or Service Acute myeloid leukemia (AML) is characterized by large numbers of abnormal, immature myeloid cells in the bone marrow and peripheral blood resulting from genetic changes in hematopoietic precursor cells which disrupt normal hematopoietic growth and differentiation (Stock, 2020). Related Policies: Genetic Cancer Susceptibility Using Next Generation Sequencing AHS-M2066 Molecular Panel Testing of Cancers to Identify Targeted Therapy AHS-M2109 Serum Tumor Markers for Malignancies AHS-G2124 Minimal Residual Disease (MRD) AHS- M2175 ***Note: This Medical Policy is complex and technical. For questions concerning the technical language and/or specific clinical indications for its use, please consult your physician. Policy BCBSNC will provide coverage for genetic testing for acute myeloid leukemia when it is determined to be medically necessary because the medical criteria and guidelines shown below are met. Benefits Application This medical policy relates only to the services or supplies described herein. Please refer to the Member's Benefit Booklet for availability of benefits. Member's benefits may vary according to benefit design; therefore member benefit language should be reviewed before applying the terms of this medical policy. When Genetic Testing for Acute Myeloid Leukemia is covered The use of genetic testing for acute myeloid leukemia is considered medically necessary for the following: A. Genetic testing for FLT3 internal tandem duplication and tyrosine kinase domain mutations (ITD and TKD), IDH1, IDH2, TET2, WT1, DNMT3A, ASXL1 and/or TP53 in adult and pediatric patients with suspected or confirmed AML of any type for prognostic and/or therapeutic purposes. -

How Close Are We to Incorporating Measurable Residual Disease Into
REVIEW ARTICLE How close are we to incorporating Ferrata Storti Foundation measurable residual disease into clinical practice for acute myeloid leukemia? Nicholas J. Short and Farhad Ravandi Department of Leukemia, The University of Texas MD Anderson Cancer Center, Houston, TX, USA Haematologica 2019 ABSTRACT Volume 104(8):1532-1541 ssessment of measurable residual disease, also called “minimal resid - ual disease,” in patients with acute myeloid leukemia in morpholog - Aical remission provides powerful prognostic information and comple - ments pretreatment factors such as cytogenetics and genomic alterations. Based on data that low levels of persistent or recurrent residual leukemia are consistently associated with an increased risk of relapse and worse long- term outcomes, its routine assessment has been recommended by some experts and consensus guidelines. In addition to providing important prog - nostic information, the detection of measurable residual disease may also theoretically help to determine the optimal post-remission strategy for an individual patient. However, the full therapeutic implications of measurable residual disease are uncertain and thus controversy exists as to whether it should be routinely incorporated into clinical practice. While some evidence supports the use of allogeneic stem cell transplantation or hypomethylating agents for some subgroups of patients in morphological remission but with detectable residual leukemia, the appropriate use of this information in mak - ing clinical decisions remains largely speculative at present. To resolve this pressing clinical issue, several ongoing studies are evaluating measurable Correspondence: residual disease-directed treatments in acute myeloid leukemia and may FARHAD RAVANDI lead to new, effective strategies for patients in these circumstances. This [email protected] review examines the common technologies used in clinical practice and in the research setting to detect residual leukemia, the major clinical studies Received: February 8, 2019. -

Regulation of the C/Ebpα Signaling Pathway in Acute Myeloid Leukemia (Review)
ONCOLOGY REPORTS 33: 2099-2106, 2015 Regulation of the C/EBPα signaling pathway in acute myeloid leukemia (Review) GUANHUA SONG1, LIn Wang2, Kehong BI3 and guosheng JIang1 1Department of hemato-oncology, Institute of Basic Medicine, shandong academy of Medical sciences, Key Laboratory for Modern Medicine and Technology of Shandong Province, Key Laboratory for Rare and Uncommon Diseases, Key Medical Laboratory for Tumor Immunology and Traditional Chinese Medicine Immunology of shandong Province, Jinan, Shandong 250062; 2Research Center for Medical Biotechnology, Shandong Academy of Medical Sciences, Jinan, Shandong 250062; 3Department of Hematology, Qianfoshan Mountain Hospital of Shandong University, Jinan, Shandong 250014, P.R. China Received December 2, 2014; Accepted January 26, 2015 DoI: 10.3892/or.2015.3848 Abstract. The transcription factor CCAAT/enhancer binding Contents protein α (C/EBPα), as a critical regulator of myeloid devel- opment, directs granulocyte and monocyte differentiation. 1. Introduction Various mechanisms have been identified to explain how 2. Function of C/EBPα in myeloid differentiation C/EBPα functions in patients with acute myeloid leukemia 3. Regulation of the C/EBPα signaling pathway (AML). C/EBPα expression is suppressed as a result of 4. Conclusion common leukemia-associated genetic and epigenetic altera- tions such as AML1-ETO, RARα-PLZF or gene promoter methylation. Recent data have shown that ubiquitination modi- 1. Introduction fication also contributes to its downregulation. In addition, 10-15% of patients with AML in an intermediate cytogenetic Acute myeloid leukemia (AML) is characterized by uncon- risk subgroup were characterized by mutations of the C/EBPα trolled proliferation of myeloid progenitors that exhibit a gene. -

Cancer and the Immune System an Overview of Recent Publications Featuring Illumina® Technology 2 Cancer and the Immune System TABLE of CONTENTS
Cancer and the Immune System An Overview of Recent Publications Featuring Illumina® Technology 2 Cancer and the Immune System TABLE OF CONTENTS 04 Introduction 05 Dendritic cells 07 T-Cell Repertoire 09 Intratumoral T-Cells 11 Single Cells and TCR Sequencing 14 Cancer antigens 18 Cancer Immunoediting 20 Tumor Microenvironment 22 Cancer Immunotherapy 25 Hematological Malignancies 28 Tracking Malignant Lymphocytes 30 Bibliography Cancer and the Immune System 3 INTRODUCTION Advances in high-throughput sequencing have dramatically improved our knowledge of the cancer genome and the intracellular mechanisms involved in tumor progression and response to treatment. While the primary focus to date has been on the cancer cell, this technology can also be used to understand the interaction of the tumor cells and the cells in the surrounding tumor microenvironment. The tumor microenvironment is defined as the cellular environment in which the tumor exists. This includes surrounding blood vessels, immune cells, fibroblasts, other cells, signaling molecules, and the extracellular matrix. Expression analysis of the RNA levels can be used to determine the activation of pathways in the tumor microenvironment. Since common signaling pathways are involved in manifestation of several hallmarks of cancer, including cancer cell proliferation, survival, invasion, metastasis, and immunosuppression, targeting these shared signaling pathways in combination with immunotherapy may be a promising strategy for cancer treatment1. It is important to note that RNA-seq has the potential to track the activation of individual clones, which could ultimately lead to personalized treatment2,3. The human adaptive immune system provides protection against an enormous variety of pathogens and well as tumors. -

Minimal Residual Disease Detection in Mantle Cell Lymphoma
Original Article Minimal residual disease detection in mantle cell lymphoma: methods and significance of four-color flow cytometry compared to consensus IGH-polymerase chain reaction at initial staging and for follow-up examinations Sebastian Böttcher,1 Matthias Ritgen,1 Sebastian Buske,1 Stefan Gesk,2 Wolfram Klapper,3 Eva Hoster,4 Wolfgang Hiddemann,4 Michael Unterhalt,4 Martin Dreyling,4 Reiner Siebert,2 Michael Kneba,1 and Christiane Pott1 on behalf of the EU MCL MRD Group 1University of Schleswig-Holstein, Campus Kiel, 2nd Department of Medicine, Kiel, Germany; 2University of Schleswig-Holstein, Campus Kiel, Institute of Human Genetics, Kiel, Germany; 3Department of Haematopathology and Lymph Node Registry Kiel, University of Schleswig-Holstein, Campus Kiel, Germany and 4Department of Internal Medicine III, University of Munich, Hospital Grosshadern, Munich, Germany Citation: Böttcher S, Ritgen M, Buske S, Gesk S, Klapper W, Hoster E, Hiddemann W, Unterhalt M, Dreyling M, Siebert R, Kneba M and Pott C on behalf of the EU MCL MRD Group. Minimal residual disease detection in mantle cell lymphoma: methods and significance of four-color flow cytometry compared to consensus IGH-polymerase chain reaction at initial staging and for follow-up examinations. Haematologica 2008 Apr; 93(4):XXX-XXX. doi: 10.3324/haematol.11267 Treatment protocols as published by the BIOMED-2 Concerted Action.3 FAM- labeled PCR products were size separated on a high-resolu- Untreated patients with Ann Arbor stage II to IV MCL tion polyacrylamide gel and laser-induced fluorescence ana- were eligible for inclusion in the trials. Computed tomogra- lyzed using an ABI 310 genetic analyzer (Applied phy (CT) examinations of neck, chest, abdomen, and pelvis Biosystems, ABI, Darmstadt, Germany) as described previ- as well as bone marrow biopsies for cytology and histology ously (GeneScanning).4 In case of a polyclonal signal after (in general assessed by local pathologists) were mandatory FR1-IGH PCR, FR2- and FR3-IGH primer sets were used in for initial staging. -

CEBPA Gene CCAAT Enhancer Binding Protein Alpha
CEBPA gene CCAAT enhancer binding protein alpha Normal Function The CEBPA gene provides instructions for making a protein called CCAAT enhancer- binding protein alpha. This protein is a transcription factor, which means that it attaches ( binds) to specific regions of DNA and helps control the activity (expression) of certain genes. CCAAT enhancer-binding protein alpha is involved in the maturation ( differentiation) of certain blood cells. It is also believed to act as a tumor suppressor, which means that it is involved in cellular mechanisms that help prevent the cells from growing and dividing too rapidly or in an uncontrolled way. Health Conditions Related to Genetic Changes Familial acute myeloid leukemia with mutated CEBPA At least six mutations in the CEBPA gene have been identified in families with familial acute myeloid leukemia with mutated CEBPA, which is a form of a blood cancer known as acute myeloid leukemia. These inherited mutations are present throughout a person' s life in virtually every cell in the body. The mutations result in a shorter version of CCAAT enhancer-binding protein alpha. This shortened protein is produced from one copy of the CEBPA gene in each cell, and it is believed to interfere with the tumor suppressor function of the normal protein produced from the second copy of the gene. Absence of the tumor suppressor function of CCAAT enhancer-binding protein alpha is believed to disrupt the regulation of blood cell production, leading to the uncontrolled production of abnormal cells that occurs in acute myeloid leukemia. In addition to the inherited mutation in one copy of the CEBPA gene in each cell, most individuals with familial acute myeloid leukemia with mutated CEBPA also acquire a mutation in the second copy of the CEBPA gene.