Microbial and Phenyl Acid Dynamics During the Start-Up Phase of Anaerobic Straw Degradation in Meso- and Thermophilic Batch Reactors
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Revised Supplement 1: Reference List for Figure 1
Revised Supplement 1: Reference list for Figure 1. Manuscript title: Process disturbances in agricultural biogas production – causes, mechanisms and effects on the biogas microbiome: A review Susanne Theuerl 1,*, Johanna Klang 1, Annette Prochnow 1,2 1 Leibniz Institute for Agricultural Engineering and Bioeconomy, Max-Exth-Allee 100, 14469 Potsdam, Germany, [email protected] (ST), [email protected] (JK), [email protected] (AP) 2 Humboldt-Universität zu Berlin, Albrecht-Daniel-Thaer-Institute for Agricultural and Horticultural Sciences, Hinter der Reinhardtstr. 6-8, 10115 Berlin, Germany * Correspondence: [email protected] Tel.: +49-331-5699-900 References of Figure 1 1Abt et al. 2010, 2Parizzi et al. 2012, 3Hahnke et al. 2016 and Tomazetto et al. 2018, 4Ueki et al. 2006 and Gronow et al. 2011, 5Grabowski et al. 2005, 6Chen and Dong 2005, 7Avgustin et al. 1997 and Purushe et al. 2010, 8Yamada et al. 2006 and Matsuura et al. 2015, 9Yamada et al. 2007 and Matsuura et al. 2015, 10Sun et al. 2016, 11Suen et al. 2011, 12Hahnke et al. 2014 and Tomazetto et al. 2016, 13Mechichi et al. 1999, 14Koeck et al. 2015a and 2015b, 15Tomazetto et al. 2017, 16Fonknechten et al. 2010, 17Chen et al. 2010, 18Nishiyama et al. 2009, 19Sieber et al. 2010, 20Plerce et al. 2008, 21Westerholm et al. 2011 and Müller et al. 2015, 22Ueki et al. 2014, 23Jackson et al. 1999 and McInerney et al. 2007, 24Ma et al. 2017, 25Harmsen et al. 1998 and Plugge et al. 2012, 26Menes and Muxi 2002, Mavromatis et al. 2013 and Hania et al. -

Lascolabacillus Massiliensis'': a New Species Isolated
NEW MICROBES IN HUMANS “Lascolabacillus massiliensis”: a new consent, and the agreement of the National Ethics Committee of Senegal and the local ethics committee of the IFR48 species isolated from the human gut (Marseille, France) were obtained under numbers 11-017 and 09-022, respectively. The initial growth was obtained after 10 days of culture in a 5% sheep blood-enriched sheep rumen M. Beye, S. Bakour, S. I. Traore, D. Raoult and P.-E. Fournier medium in aerobic atmosphere at 37°C. The bacterium was Unité de Recherche en Maladies Infectieuses et Tropicales Emergentes, sub-cultured on 5% sheep blood-enriched Columbia agar Institut Hospitalo-Universitaire Méditerranée-Infection, Aix-Marseille (bioMérieux, Marcy l’Etoile, France) and grew in 24 hours at Université, Faculté de Médecine, Marseille cedex 5, France 37°C in both aerobic and anaerobic conditions. Agar-grown colonies were pale grey and 1.5 mm in diameter. Bacterial cells were Gram-negative, rod-shaped and polymorphic, ranginginlengthfrom1.5to10μm. Strain SIT8 was catalase- Abstract and oxidase-negative. The 16S rRNA gene was sequenced using the fD1-rP2 primers as previously described, using a 3130-XL sequencer (Applied Biosciences, Saint Aubin, We report here the main characteristics of “Lascolabacillus France). Strain SIT8 exhibited a 94.14% sequence identity with massiliensis” strain SIT8 (CSUR P1560) that was isolated from the Proteiniphilum acetatigenes strain TB107 (GenBank accession stool of a healthy 28-month-old boy. NR_043154), the phylogenetically closest species with New Microbes and New Infections © 2016 The Authors. Published standing in nomenclature (Fig. 1), which putatively classifies it by Elsevier Ltd on behalf of European Society of Clinical as a member of a new genus within the family Porphyr- Microbiology and Infectious Diseases. -
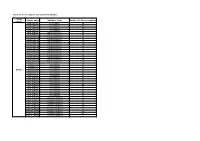
Table S1: List of Samples Included in the Analysis
Table S1: list of samples included in the analysis Study Sample name Inhibitory status Number of days at sampling number DNA.0P2T4 No inhibition 29 DNA.0P2T6 No inhibition 57 DNA.10P2T4 No inhibition 29 DNA.10P2T6 No inhibition 57 DNA.75P2T4 Phenol inhibition 29 DNA.75P2T6 Phenol inhibition 57 DNA.100P2T4 Phenol inhibition 29 DNA.100P2T6 Phenol inhibition 57 DNA.125P1T4 Phenol inhibition 29 DNA.125P1T6 Phenol inhibition 57 DNA.125P2T4 Phenol inhibition 29 DNA.125P2T6 Phenol inhibition 57 DNA.125P3T4 Phenol inhibition 29 DNA.125P3T6 Phenol inhibition 57 DNA.150P2T4 Phenol inhibition 29 DNA.150P2T6 Phenol inhibition 57 DNA.200P2T4 Phenol inhibition 29 DNA.200P2T6 Phenol inhibition 57 DNA.0N2T4 No inhibition 29 DNA.0N2T5 No inhibition 42 DNA.0N2T6 No inhibition 57 Study 1 DNA.5N2T4 No inhibition 29 DNA.5N2T5 No inhibition 42 DNA.5N2T6 No inhibition 57 DNA.10N2T4 No inhibition 29 DNA.10N2T5 No inhibition 42 DNA.10N2T6 No inhibition 57 DNA.15N2T4 No inhibition 29 DNA.15N2T5 No inhibition 42 DNA.15N2T6 No inhibition 57 DNA.25N2T4 No inhibition 29 DNA.25N2T5 No inhibition 42 DNA.25N2T6 No inhibition 57 DNA.75N2T4 Ammonia inhibition 29 DNA.75N2T5 Ammonia inhibition 42 DNA.75N2T6 Ammonia inhibition 57 DNA.100N2T4 Ammonia inhibition 29 DNA.100N2T5 Ammonia inhibition 42 DNA.100N2T6 Ammonia inhibition 57 DNA.250N2T4 Ammonia inhibition 29 DNA.250N2T5 Ammonia inhibition 42 DNA.250N2T6 Ammonia inhibition 57 nono2T3 No inhibition 16 noN2T4 Ammonia inhibition 23 noN2T8 Ammonia inhibition 60 noN2T9 Ammonia inhibition 85 noPhi2T4 Phenol inhibition 23 noPhi2T5 -

Characterization of Cucumber Fermentation Spoilage Bacteria by Enrichment Culture and 16S Rdna Cloning
Characterization of Cucumber Fermentation Spoilage Bacteria by Enrichment Culture and 16S rDNA Cloning Fred Breidt, Eduardo Medina, Doria Wafa, Ilenys P´erez-D´ıaz, Wendy Franco, Hsin-Yu Huang, Suzanne D. Johanningsmeier, and Jae Ho Kim Abstract: Commercial cucumber fermentations are typically carried out in 40000 L fermentation tanks. A secondary fermentation can occur after sugars are consumed that results in the formation of acetic, propionic, and butyric acids, concomitantly with the loss of lactic acid and an increase in pH. Spoilage fermentations can result in significant economic loss for industrial producers. The microbiota that result in spoilage remain incompletely defined. Previous studies have implicated yeasts, lactic acid bacteria, enterobacteriaceae, and Clostridia as having a role in spoilage fermentations. We report that Propionibacterium and Pectinatus isolates from cucumber fermentation spoilage converted lactic acid to propionic acid, increasing pH. The analysis of 16S rDNA cloning libraries confirmed and expanded the knowledge gained from previous studies using classical microbiological methods. Our data show that Gram-negative anaerobic bacteria supersede Gram-positive Fermincutes species after the pH rises from around 3.2 to pH 5, and propionic and butyric acids are produced. Characterization of the spoilage microbiota is an important first step in efforts to prevent cucumber fermentation spoilage. Keywords: pickled vegetables, Pectinatus, Propionibacteria, secondary cucumber fermentation, spoilage M: Food Microbiology Practical Application: An understanding of the microorganisms that cause commercial cucumber fermentation spoilage & Safety may aid in developing methods to prevent the spoilage from occurring. Introduction cucumbers fermented at 2.3% NaCl (Fleming and others 1989). Commercial cucumber fermentations are typically carried out In this fermentation tank, the initial lactic acid fermentation was in large 40000 L outdoor tanks (reviewed by Breidt and others completed within 2 wk, with 1.2% lactic acid formed (pH 3.6) 2007). -

Microbial Communities Associated with Stable Fly (Diptera: Muscidae) Larvae and Their Developmental Substrates Erin Scully USDA-ARS, [email protected]
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Faculty Publications: Department of Entomology Entomology, Department of 2017 Microbial Communities Associated With Stable Fly (Diptera: Muscidae) Larvae and Their Developmental Substrates Erin Scully USDA-ARS, [email protected] Kristina Friesen USDA-ARS, [email protected] Brian Wienhold USDA-ARS, [email protected] Lisa M. Durso USDA-ARS, [email protected] Follow this and additional works at: http://digitalcommons.unl.edu/entomologyfacpub Part of the Entomology Commons Scully, Erin; Friesen, Kristina; Wienhold, Brian; and Durso, Lisa M., "Microbial Communities Associated With Stable Fly (Diptera: Muscidae) Larvae and Their eD velopmental Substrates" (2017). Faculty Publications: Department of Entomology. 502. http://digitalcommons.unl.edu/entomologyfacpub/502 This Article is brought to you for free and open access by the Entomology, Department of at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Faculty Publications: Department of Entomology by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. Annals of the Entomological Society of America, 110(1), 2017, 61–72 doi: 10.1093/aesa/saw087 Special Collection: Filth Fly–Microbe Interactions Research article Microbial Communities Associated With Stable Fly (Diptera: Muscidae) Larvae and Their Developmental Substrates Erin Scully,1 Kristina Friesen,2,3 Brian Wienhold,2 and Lisa M. Durso2 1USDA, ARS, Stored Product -

Page 1 of 41 RSC Advances
RSC Advances This is an Accepted Manuscript, which has been through the Royal Society of Chemistry peer review process and has been accepted for publication. Accepted Manuscripts are published online shortly after acceptance, before technical editing, formatting and proof reading. Using this free service, authors can make their results available to the community, in citable form, before we publish the edited article. This Accepted Manuscript will be replaced by the edited, formatted and paginated article as soon as this is available. You can find more information about Accepted Manuscripts in the Information for Authors. Please note that technical editing may introduce minor changes to the text and/or graphics, which may alter content. The journal’s standard Terms & Conditions and the Ethical guidelines still apply. In no event shall the Royal Society of Chemistry be held responsible for any errors or omissions in this Accepted Manuscript or any consequences arising from the use of any information it contains. www.rsc.org/advances Page 1 of 41 RSC Advances 1 Regulation of acidogenic metabolism towards enhanced short chain fatty acids biosynthesis 2 from waste: Metagenomic Profiling 3 4 Omprakash Sarkar, A. Naresh Kumar, Shikha Dahiya, K.Vamshi Krishna, Dileep Kumar 5 Yeruva, S.Venkata Mohan* 6 Bioengineering and Environmental Sciences (BEES), CSIR-Indian Institute of Chemical 7 Technology (CSIR-IICT), Hyderabad 500 007, India 8 *E-mail: [email protected]; Tel: 0091-40-27161765 9 10 Abstract 11 Short chain carboxylic (volatile fatty) acids (VFA) production in mixed microbiomes is majorly 12 limited by the prevalence of the methanogenic bacteria and availability of substrate from waste 13 to the biocatalyst during the fermentation process. -

Proteiniborus Ethanoligenes Gen. Nov., Sp. Nov., an Anaerobic Protein-Utilizing Bacterium
%paper no. ije65108 charlesworth ref: ije65108& New Taxa - Other Gram-positive Bacteria International Journal of Systematic and Evolutionary Microbiology (2008), 58, 000–000 DOI 10.1099/ijs.0.65108-0 Proteiniborus ethanoligenes gen. nov., sp. nov., an anaerobic protein-utilizing bacterium Lili Niu,1,2 Lei Song1,2 and Xiuzhu Dong1 Correspondence 1State Key Laboratory of Microbial Resources, Institute of Microbiology, Chinese Academy of Xiuzhu Dong Sciences, Beijing 100101, PR China [email protected] 2Graduate School, Chinese Academy of Sciences, Beijing 100049, PR China A novel anaerobic, mesophilic, protein-utilizing bacterial strain, GWT, was isolated from the mesophilic hydrogen-producing granular sludge used to treat food industry wastewater. The strain was a Gram-positive, non-spore-forming and non-motile rod. Growth of the strain was observed at 20–48 6C and at pH 6.4–10.0. The strain used yeast extract and peptone as carbon and energy sources. Weak growth was also observed with tryptone and Casamino acids as carbon and energy sources. The strain used none of the tested carbohydrates, alcohols or fatty acids. The fermentation products in peptone-yeast broth included ethanol, acetic acid, hydrogen and carbon dioxide. Gelatin was not hydrolysed. Nitrate was reduced. Indole was produced. T NH3 and H2S were not produced. The DNA G+C content of strain GW was 38.0 mol%. The predominant cellular fatty acids were the saturated fatty acids C14 : 0 (15.58 %), C16 : 0 (25.40 %) and C18 : 0 (12.03 %). Phylogenetic analysis based on 16S rRNA gene sequence similarity revealed that strain GWT represented a new branch within cluster XII of the Clostridium subphylum, with ,89.6 % 16S rRNA gene sequence similarities to all described species. -

Draft Genome Sequence of Fermentimonas Caenicola Strain SIT8, Isolated from the Human
Beye et al. Standards in Genomic Sciences (2018) 13:8 https://doi.org/10.1186/s40793-018-0310-6 EXTENDED GENOME REPORT Open Access Draft genome sequence of Fermentimonas caenicola strain SIT8, isolated from the human gut Mamadou Beye1, Sofiane Bakour1, Sory Ibrahima Traore1, Jaishriram Rathored1, Noémie Labas1, Didier Raoult1,2 and Pierre-Edouard Fournier1* Abstract We report the properties of a draft genome sequence of the bacterium Fermentimonas caenicola strain SIT8 (= CSUR P1560). This strain, whose genome is described here, was isolated from the fecal flora of a healthy 28-month-old Senegalese boy. Strain SIT8 is a facultatively anaerobic Gram-negative bacillus. Here we describe the features of this organism, together with the complete genome sequence and annotation. The 2,824,451-bp long (1 chromosome but no plasmid) contains 2354 protein-coding and 46 RNA genes, including four rRNA genes. Keywords: Fermentimonas caenicola,Genome,Humangut,Microbiota Introduction Herein, we present a set of features for F. caenicola Fermentimonas caenicola strain SIT8 (= CSUR P1560) strain SIT8 together with the description of the was isolated from the stool of a healthy 28-month-old complete genomic sequence and annotation. Senegalese boy as part of a study aiming at cultivating all species within the human gastro-intestinal micro- Organism information biota. It is a Gram-negative, facultatively anaerobic, Classification and features indole-negative bacillus. Initially, we had named this Fermentimonas caenicola “Lascolabacillus massiliensis” strain SIT8 was isolated from bacterium as it exhibited the stool of a healthy 28-month-old Senegalese boy unique features among members of the family Porphyro- ’ monadaceae (Table 1). -

Supplementary Material Evidence of Spatial Homogeneity in an Electromethanogenic Cathodic Microbial Community
Evidence of Spatial Homogeneity in an Electromethanogenic Cathodic Microbial Community. Item Type Article Authors Ragab, Alaa I.; Katuri, Krishna; Ali, Muhammad; Saikaly, Pascal Citation Ragab, A., Katuri, K. P., Ali, M., & Saikaly, P. E. (2019). Evidence of Spatial Homogeneity in an Electromethanogenic Cathodic Microbial Community. Frontiers in Microbiology, 10. doi:10.3389/ fmicb.2019.01747 Eprint version Publisher's Version/PDF DOI 10.3389/fmicb.2019.01747 Publisher Frontiers Media SA Journal Frontiers in microbiology Rights Ragab, Katuri, Ali and Saikaly. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms. Download date 26/09/2021 21:00:48 Item License https://creativecommons.org/licenses/by/4.0/ Link to Item http://hdl.handle.net/10754/656542 Supplementary Material Evidence of spatial homogeneity in an electromethanogenic cathodic microbial community Ala’a Ragab1, Krishna P. Katuri1, Muhammad Ali1, Pascal E. Saikaly1* 1 Biological and Environmental Science and Engineering Division, Water Desalination and Reuse Center, King Abdullah University of Science and Technology (KAUST), Saudi Arabia * Correspondence: Pascal E. Saikaly [email protected] Supplementary Material Figure S1. Rarefaction curves for all retrieved reads for the 263 operational taxonomic units (OTUs) for all samples faceted by reactors and combined together. Data points are colored by sampling position (top, center, bottom). -

Integrating Mineral Wastes in the Anaerobic Digestion of OFMSW for Improved Recovery of Renewable Energy
Integrating mineral wastes in the anaerobic digestion of OFMSW for improved recovery of renewable energy Burhan Shamurad [email protected] March 2020 A thesis submitted for the degree Doctor of Philosophy in the School of Engineering, Newcastle University, UK ii Abstract This thesis investigated the effects of mineral wastes (MW) on laboratory-scale anaerobic reactors treating organic wastes. Different MW resources were used, incineration bottom ash (IBA), fly ash (FA) and boiler ash (BA), taken from a municipal solid waste incineration (MSWI) plants, as well as a cement-based waste (CBW) from construction demolition wastes. The hypothesis was that these MW would provide trace elements (TEs) deficient in the organic fraction of municipal solid waste (OFMSW), and offer moderate alkalinity to prevent reactor acidification of mesophilic anaerobic digestion of the OFMSW. The control and operation of batch biomethane potential (BMP) reactors and continuous stirred tank reactors (CSTRs; single-stage and two-stage reactors), was studied under different feeding regimes, different organic loading rates and hydraulic/solid retention times, in order to determine potential benefits of mineral waste amendments and aqueous trace metal supplements on anaerobic digestion efficiency, methane productivity and process stability. Co-digestion of different solid MW and organic wastes in single-stage CSTR using a liquid- recycled feeding method (LRFM) enhanced process stability (pH of 6.8 – 7.2), increased methane production by 25 - 45%, and yielded 450 – 520 mL CH4/g VS (near to the theoretical maximum) compared to the control. Whereas draw-and-fill feeding method (DFFM) also enhanced digestibility but to a lesser degree. Pre-treatment of the OFMSW with the MW at 37oC improved substrate hydrolysis, and enhanced the performance and stability of the DFFM digestion processes further to values similar to those of LRFM reactors. -

Genomic Characterization of the Uncultured Bacteroidales Family S24-7 Inhabiting the Guts of Homeothermic Animals Kate L
Ormerod et al. Microbiome (2016) 4:36 DOI 10.1186/s40168-016-0181-2 RESEARCH Open Access Genomic characterization of the uncultured Bacteroidales family S24-7 inhabiting the guts of homeothermic animals Kate L. Ormerod1, David L. A. Wood1, Nancy Lachner1, Shaan L. Gellatly2, Joshua N. Daly1, Jeremy D. Parsons3, Cristiana G. O. Dal’Molin4, Robin W. Palfreyman4, Lars K. Nielsen4, Matthew A. Cooper5, Mark Morrison6, Philip M. Hansbro2 and Philip Hugenholtz1* Abstract Background: Our view of host-associated microbiota remains incomplete due to the presence of as yet uncultured constituents. The Bacteroidales family S24-7 is a prominent example of one of these groups. Marker gene surveys indicate that members of this family are highly localized to the gastrointestinal tracts of homeothermic animals and are increasingly being recognized as a numerically predominant member of the gut microbiota; however, little is known about the nature of their interactions with the host. Results: Here, we provide the first whole genome exploration of this family, for which we propose the name “Candidatus Homeothermaceae,” using 30 population genomes extracted from fecal samples of four different animal hosts: human, mouse, koala, and guinea pig. We infer the core metabolism of “Ca. Homeothermaceae” to be that of fermentative or nanaerobic bacteria, resembling that of related Bacteroidales families. In addition, we describe three trophic guilds within the family, plant glycan (hemicellulose and pectin), host glycan, and α-glucan, each broadly defined by increased abundance of enzymes involved in the degradation of particular carbohydrates. Conclusions: “Ca. Homeothermaceae” representatives constitute a substantial component of the murine gut microbiota, as well as being present within the human gut, and this study provides important first insights into the nature of their residency. -

Supplementary Materials
Supplementary Materials Effects of antibiotics on the bacterial community,metabolic functions and antibiotic resistance genes in mariculture sediments during enrichment culturing Meng-Qi Ye,1 Guan-Jun Chen1,2 and Zong-Jun Du *1,2 1 Marine College, Shandong University, Weihai, Shandong, 264209, China. 2State Key Laboratory of Microbial Technology, Shandong University, Qingdao, Shandong, 266237, China Authors for correspondence: Zong-Jun Du, Email: [email protected] Supplementary Tables Table S1 The information of antibiotics used in this study. Antibiotic name Class CAS NO. Inhibited pathway Mainly usage Zinc bacitracin Peptides 1405-89-6 Cell wall synthesis veterinary Ciprofloxacin Fluoroquinolone 85721-33-1 DNA synthesis veterinary and human Ampicillin sodium β-lactams 69-57-8 Cell wall synthesis veterinary and human Chloramphenicol Chloramphenicols 56-75-7 Protein synthesis veterinary and human Macrolides Tylosin 1401-69-0 Protein synthesis veterinary Tetracycline Tetracyclines 60-54-8 Aminoacyl-tRNA access veterinary and human Table S2 The dominant phyla (average abundance > 1.0%) in each treatment Treatment Dominant phyla Control (A) Proteobacteria (43.01%), Bacteroidetes(18.67%), Fusobacteria (12.91%), Firmicutes (11.74%), Chloroflexi (1.80%), Spirochaetae (1.64%), Planctomycetes (1.48%), Cloacimonetes (1.34%), Latescibacteria (1.03%) P Proteobacteria(50.24%), Bacteroidetes(19.79%), Fusobacteria (12.73%), Firmicutes (8.54%), Spirochaetae(2.92%), Synergistetes (1.98%), Q Proteobacteria (40.61%), Bacteroidetes (22.39%), Fusobacteria