New Insights on the Zeugodacus Cucurbitae (Coquillett) Bacteriome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Potential of a Fly Gut Microbiota Incorporated Gel-Based Larval Diet for Rearing Bactrocera Dorsalis (Hendel) Mahfuza Khan1*, Kajla Seheli1, Md
Khan et al. BMC Biotechnology 2019, 19(Suppl 2):94 https://doi.org/10.1186/s12896-019-0580-0 RESEARCH Open Access Potential of a fly gut microbiota incorporated gel-based larval diet for rearing Bactrocera dorsalis (Hendel) Mahfuza Khan1*, Kajla Seheli1, Md. Abdul Bari1, Nahida Sultana1, Shakil Ahmed Khan1, Khandokar Fahmida Sultana2 and Md. Anwar Hossain3 Abstract Background: The Oriental fruit fly, Bactrocera dorsalis (Hendel) (Diptera: Tephritidae), is an important polyphagous pest of horticultural produce. The sterile insect technique (SIT) is a proven control method against many insect pests, including fruit flies, under area-wide pest management programs. High quality mass-rearing process and the cost-effective production of sterile target species are important for SIT. Irradiation is reported to cause severe damage to the symbiotic community structure in the mid gut of fruit fly species, impairing SIT success. However, studies have found that target-specific manipulation of insect gut bacteria can positively impact the overall fitness of SIT-specific insects. Results: Twelve bacterial genera were isolated and identified from B. dorsalis eggs, third instars larval gut and adults gut. The bacterial genera were Acinetobacter, Alcaligenes, Citrobacter, Pseudomonas, Proteus, and Stenotrophomonas, belonging to the Enterobacteriaceae family. Larval diet enrichment with the selected bacterial isolate, Proteus sp. was found to improve adult emergence, percentage of male, and survival under stress. However, no significant changes were recorded in B. dorsalis egg hatching, pupal yield, pupal weight, duration of the larval stage, or flight ability. Conclusions: These findings support the hypothesis that gut bacterial isolates can be used in conjunction with SIT. -

2013. Tucuman 3Rd. RCM. August-2013. Produced Working
1 IAEA-D41023-CR-3 LIMITED DISTRIBUTION WORKING MATERIAL RESOLUTION OF CRYPTIC SPECIES COMPLEXES OF TEPHRITID PESTS TO OVERCOME CONSTRAINTS TO SIT APPLICATION AND INTERNATIONAL TRADE THIRD RESEARCH COORDINATION MEETING OF A FAO/IAEA COORDINATED RESEARCH PROJECT HELD IN TUCUMAN, ARGENTINA FROM 26-31 AUGUST 2013 Reproduced by the IAEA Vienna, Austria 2014 __________________________________________________________________________ NOTE Material in this document has been supplied by the authors and has not been edited by the IAEA. The views expressed remain the responsibility of the named authors and do not necessarily reflect those of the government of the designating Member State(s). In particular, neither the IAEA nor any other organization or body sponsoring the meeting can be held responsible for any material reproduced in this document. 2 Table of Contents A. Background Situation Analysis ................................................................................. 3 B. The Co-ordinated Research Project (CRP) ................................................................ 4 C. Report for the 3rd RCM (Tucuman 2013) ................................................................. 5 D. Conclusions on Current Status and Recommended Future Activities for the CRP Participants ................................................................................................. 10 Anastrepha fraterculus Complex ............................................................................ 10 Background Situation Analysis .................................................................. -

Indoor Microbiome, Environmental Characteristics and Asthma Among Junior High
Indoor microbiome, environmental characteristics and asthma among junior high school students in Johor Bahru, Malaysia Xi Fu1, Dan Norbäck2, Qianqian Yuan3,4, Yanling Li3,4, Xunhua Zhu3,4,Yiqun Deng3,4, Jamal Hisham Hashim5,6, Zailina Hashim7, Yi-Wu Zheng8, Xu-Xin Lai8, Michael Dho Spangfort8, Yu Sun3,4 * 1Department of Occupational and Environmental Health, School of Public Health, Sun Yat-sen University, Guangzhou, PR China. 2Occupational and Environmental Medicine, Dept. of Medical Science, University Hospital, Uppsala University, 75237 Uppsala, Sweden. 3Guangdong Provincial Key Laboratory of Protein Function and Regulation in Agricultural Organisms, College of Life Sciences, South China Agricultural University, Guangzhou, Guangdong, 510642, PR China. 4Key Laboratory of Zoonosis of Ministry of Agriculture and Rural Affairs, South China Agricultural University, Guangzhou, Guangdong, 510642, PR China. 5United Nations University-International Institute for Global Health, Kuala Lumpur, Malaysia, 6Department of Community Health, National University of Malaysia, Kuala Lumpur, Malaysia 7Department of Environmental and Occupational Health, Faculty of Medicine and Health Sciences, Universiti Putra Malaysia, UPM, Serdang, Selangor, Malaysia 8Asia Pacific Research, ALK-Abello A/S, Guanzhou, China *To whom corresponds should be addressed: Yu Sun [email protected], Key words: bacteria, fungi, microbial community, absolute quantity, wheezing, breathlessness, adolescence, dampness/visible mold Abstract Indoor microbial diversity and composition are suggested to affect the prevalence and severity of asthma, but no microbial association study has been conducted in tropical countries. In this study, we collected floor dust and environmental characteristics from 21 classrooms, and health data related to asthma symptoms from 309 students, in junior high schools in Johor Bahru, Malaysia. -
FFN41-Dec2020
FRUIT FLY TEPHRITID WORKERS DATABASE TWD NEWS FRUIT FLY NEWS FFN#41 December 2020 Fruit Fly News Guest of Honour Prof. KENG HONG TAN Guest of Honour: Professor Keng Hong Tan Prof Keng Hong Tan ("Hong" as his friends called him), is the Guest of Honor of the Fruit Fly News (FFN#41_December 2020). He agreed to share with us his walk of life, which took him to many places in the world and enabled him to meet many fruit fly workers. He kindly agreed to share his experience and some of his personal photos. Read more New Fruit Fly Projects National Fruit Fly Strategy 2020–2025 FruitFlyNet-II The Australian National Fruit Fly Strategy 2020– FruitFlyNet-ii is a Strategic Project for Open 2025 (NFFS) has been developed by the National Innovation funded by ENICBCMED/EU program. Fruit Fly Council (the Council) to provide a It involves 5 countries: Greece, Spain, Italy, framework for ongoing stakeholder cooperation Lebanon and Tunisia. The project aims to develop to support a contemporary, viable, cost-effective for the first time a fully automated e-monitoring, and coordinated national approach to fruit fly IPM spraying, e-solutions and environmental management. Read more friendly control of the olive fruit fly and the Medfly. Read more Project F3 Fruit Fly Free Fruit Fly IPM-Horizon 2020 South Africa, Mozambique and Belgium started a The Fruit fly Integrated Pest Management project, new project funded by the World Trade funded by the European Commission, shows Organization through their STDF programme. resilience and innovation during Covid-19 times. The objective is to develop a regionally Despite the obstacles, FF-IPM’s researchers harmonised framework for development and managed to produce high-quality results and held implementation of recognised Pest Free Areas virtual meetings and workshops. -

A Survey of Fruit-Feeding Insects and Their Parasitoids Occurring on Wild Olives, Olea Europaea Ssp
This article was downloaded by: [USDA National Agricultural Library] On: 11 February 2009 Access details: Access Details: [subscription number 790740294] Publisher Taylor & Francis Informa Ltd Registered in England and Wales Registered Number: 1072954 Registered office: Mortimer House, 37-41 Mortimer Street, London W1T 3JH, UK Biocontrol Science and Technology Publication details, including instructions for authors and subscription information: http://www.informaworld.com/smpp/title~content=t713409232 A survey of fruit-feeding insects and their parasitoids occurring on wild olives, Olea europaea ssp. cuspidata, in the Eastern Cape of South Africa Nolwazi Mkize a; Kim A. Hoelmer b; Martin H. Villet a a Department of Zoology & Entomology, Rhodes University, Grahamstown, South Africa b United States Department of Agriculture, Agricultural Research Service, Beneficial Insect Introduction Research Unit, Newark, USA Online Publication Date: 01 January 2008 To cite this Article Mkize, Nolwazi, Hoelmer, Kim A. and Villet, Martin H.(2008)'A survey of fruit-feeding insects and their parasitoids occurring on wild olives, Olea europaea ssp. cuspidata, in the Eastern Cape of South Africa',Biocontrol Science and Technology,18:10,991 — 1004 To link to this Article: DOI: 10.1080/09583150802450154 URL: http://dx.doi.org/10.1080/09583150802450154 PLEASE SCROLL DOWN FOR ARTICLE Full terms and conditions of use: http://www.informaworld.com/terms-and-conditions-of-access.pdf This article may be used for research, teaching and private study purposes. Any substantial or systematic reproduction, re-distribution, re-selling, loan or sub-licensing, systematic supply or distribution in any form to anyone is expressly forbidden. The publisher does not give any warranty express or implied or make any representation that the contents will be complete or accurate or up to date. -

Title Floral Synomone Diversification of Sibling Bulbophyllum Species
Floral synomone diversification of sibling Bulbophyllum Title species (Orchidaceae) in attracting fruit fly pollinators Nakahira, Masataka; Ono, Hajime; Wee, Suk Ling; Tan, Keng Author(s) Hong; Nishida, Ritsuo Citation Biochemical Systematics and Ecology (2018), 81: 86-95 Issue Date 2018-12 URL http://hdl.handle.net/2433/235528 © 2018. This manuscript version is made available under the CC-BY-NC-ND 4.0 license http://creativecommons.org/licenses/by-nc-nd/4.0/.; The full- text file will be made open to the public on 01 December 2019 Right in accordance with publisher's 'Terms and Conditions for Self- Archiving'.; This is not the published version. Please cite only the published version. この論文は出版社版でありません。 引用の際には出版社版をご確認ご利用ください。 Type Journal Article Textversion author Kyoto University Floral Synomone Diversification of Bulbophyllum Sibling Species (Orchidaceae) in Attracting Fruit Fly Pollinators Masataka Nakahiraa · Hajime Onoa · Suk Ling Weeb,c · Keng Hong Tand · Ritsuo Nishidaa, * *Corresponding author Ritsuo Nishida [email protected] a Laboratory of Chemical Ecology, Graduate School of Agriculture, Kyoto University, Kyoto 606- 8502, Japan b School of Environmental and Natural Resource Sciences, Faculty of Science and Technology, Universiti Kebangsaan Malaysia, 43600 Bangi Selangor Darul Ehsan, Malaysia c Centre for Insect Systematics, Faculty of Science and Technology, Universiti Kebangsaan Malaysia, 43600 Bangi Selangor Darul Ehsan, Malaysia d Tan Hak Heng Co., Johor Bahru, Johor, Malaysia 1 Abstract Floral scent is one of the crucial cues to attract specific groups of insect pollinators in angiosperms. We examined the semiochemical diversity in the interactions between “fruit fly orchids” and their pollinator fruit fly species in two genera, Bactrocera and Zeugodacus (Tephritidae: Diptera). -

Parasitoids of Queensland Fruit Fly Bactrocera Tryoni in Australia and Prospects for Improved Biological Control
Insects 2012, 3, 1056-1083; doi:10.3390/insects3041056 OPEN ACCESS insects ISSN 2075-4450 www.mdpi.com/journal/insects/ Review Parasitoids of Queensland Fruit Fly Bactrocera tryoni in Australia and Prospects for Improved Biological Control Ashley L. Zamek 1,, Jennifer E. Spinner 2 Jessica L. Micallef 1, Geoff M. Gurr 3 and Olivia L. Reynolds 4,* 1 Elizabeth Macarthur Agricultural Institute, NSW Department of Primary Industries, Woodbridge Road, Menangle, NSW 2568, Australia; E-Mails: [email protected] (A.L.Z.); [email protected] (J.L.M) 2 EH Graham Centre for Agricultural Innovation, NSW Department of Primary Industries and Charles Sturt University, Locked Bag 588, Wagga Wagga, NSW 2678, Australia; E-Mail: [email protected] 3 EH Graham Centre for Agricultural Innovation, NSW Department of Primary Industries and Charles Sturt University, Charles Sturt University, P.O. Box 883, Orange, NSW 2800, Australia; E-Mail: [email protected] 4 EH Graham Centre for Agricultural Innovation, NSW Department of Primary Industries and Charles Sturt University, Elizabeth Macarthur Agricultural Institute, Woodbridge Road, Menangle, NSW 2568, Australia Present address: Level 1, 1 Phipps Close DEAKIN ACT 2600 Australia. * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +61-0-2-4640-6426; Fax: +61-0-2-4640-6300. Received: 3 September 2012; in revised form: 4 October 2012 / Accepted: 10 October 2012 / Published: 22 October 2012 Abstract: This review draws together available information on the biology, methods for study, and culturing of hymenopteran parasitoids of the Queensland fruit fly, Bactrocera tryoni, and assesses prospects for improving biological control of this serious pest. -

The Sterile Insect Technique for Control of the Oriental Fruit Fly, Bactrocera Dorsalis (Hendel), in Mango Orchards in Ratchaburi Province, Thailand
Proceedings of 6th International Fruit Fly Symposium 6–10 May 2002, Stellenbosch, South Africa pp. 223–232 The sterile insect technique for control of the oriental fruit fly, Bactrocera dorsalis (Hendel), in mango orchards in Ratchaburi Province, Thailand M. Sutantawong1, W. Orankanok2, W.R. Enkerlin3*, V. Wornoayporn4 & C. Caceres4 1Office of Atomic Energy for Peace, Thailand 2Institute of Irradiation for Agricultural Development, Department of Agricultural Extension, Thailand 3Insect Pest Control Section, Joint FAO/IAEA Division in Food and Agriculture, Wagramer Strasse 5, P.O. Box 100, A-1400 Vienna, Austria 4Entomology Unit, FAO/IAEA Agriculture and Biotechnology Laboratory, Seibersdorf, Austria Fruit flies are the main constraint to improving production and trade of fruits and vegetables in Thailand. Therefore, since 1987, the Department of Agricultural Extension (DOAE) in cooperation with the Office of Atomic Energy for Peace has run a pilot project for control of the oriental fruit fly (OFF),Bactrocera dorsalis (Hendel),by integrating the sterile insect technique (SIT) with other moni- toring and control methods in the mango-production areas in the Paktor District in the Ratchaburi province. The project includes mass-rearing and sterilization of OFF at the mass-rearing and steril- ization facility of the Irradiation for Agricultural Development Institute, DOAE, located in the Pathumthani Province and field releases of sterile flies complemented by bait sprays and a monitor- ing network of methyl eugenol baited traps. The International Atomic Energy Agency has pro- vided technical assistance since 1991 through a Technical Cooperation Project. The assistance has resulted in improved rearing and field operation activities. In the years 1999 and 2000, weekly ground shipments of 5–10 million sterile pupae were transported from the production facility in Pathumthani to Paktor District for ground release in 1120 ha of small commercial mango orchards. -

(Diptera: Tephritidae) Control in Mango
OPTIMIZATION OF AN INTEGRATED PEST MANAGEMENT PROGRAM FOR FRUIT FLIES (DIPTERA: TEPHRITIDAE) CONTROL IN MANGO: A CASE OF MANICA PROVINCE, MOZAMBIQUE LAURA DA GRAҪA JOSÉ CANHANGA A THESIS SUBMITTED IN FULFILMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY OF SOKOINE UNIVERSITY OF AGRICULTURE. MOROGORO, TANZANIA. 2018 ii EXTENDED ABSTRACT This study was undertaken to reduce the losses caused by Bactrocera dorsalis (Hendel) in Manica province, Mozambique, through an optimized integrated pest management (IPM) package. It involved interviews with farmers to collect baseline information on awareness of fruit producers regarding fruit fly pests and their management so that an IPM package can be developed based on the farmers’ needs. Additionally, systematic trapping data of B. dorsalis seasonality and damage were collected and economic injury level (EIL) for B. dorsalis was estimated. Based on EIL, the IPM for B. dorsalis control developed in Tanzania by the Sokoine University of Agriculture (SUA IPM) was optimized. The SUA IPM included calendar GF 120 NF bait sprays and orchard sanitation while for the optimized IPM the GF 120 NF was only sprayed in the subplots inside the orchard when the threshold of 30 flies/trap/week was reached. The effectiveness of SUA IPM and its optimized version were also tested. Results showed that fruit flies were the main pest problem in mango and citrus orchards. More than 70% the respondents indicated low fruit quality and increasing volumes of uncommercial zed fruits as consequences of fruit flies infestation. The monetary value of losses reached a value of USD 135,784.8 during 2014/15 mango season. -

Life History of the Melon Fly
LIFE HISTORY OF THE MELON FLY By E. A. BACK, Entomological assistant, and C. E. PEMBëRTON, Scientific Assistant, Mediterranean Fruit-Fly Investigations, Bureau of Entomology INTRODUCTION Aside from the Mediterranean fruit fly, Ceratitis capitata Wied., there is no other insect in the Hawaiian Islands that is causing such financial loss to fruit and vegetable interests as the melon fly, Bactrocera cucurbitae Coq. The damages caused by its ravages are placed by some even higher than those caused by C. capitata. While B. cucurbitae was not officially recorded until November, 1898, when it was first discovered by Mr. George Compere in the market gardens in the environs of Honolulu, it had been knownlocally about that city many years before. Mr. Albert Waterhouse, Acting President of the Hawaiian Board of Agriculture and Forestry, states that less than 30 years ago excellent cantaloupes (Cucumis meló) and watermelons (Citrullus vulgaris) and many kinds of pumpkins and squashes were grown in profusion the year round. Since that time the spread of the melon fly has been so rapid that this insect is now found on all the important islands of the Hawaiian group, and cantaloupes and water- melons can not be grown except on new land distant from old gardens. More than 95 per cent of the pumpkin (Cucúrbita pepo) crop is annually ruined, not to mention the havoc caused among the more resistant cucumbers (Cucumis sativus). ^ Not only does the adult melon fly oviposit in fruit that has already set, but more often—in the case of the pumpkin and the squash (Cucúrbita spp.)—in the unopened male and female flowers, in the stem of the vine, and even in the seedling itself, especially in seedlings of the watermelon and the cantaloupe. -
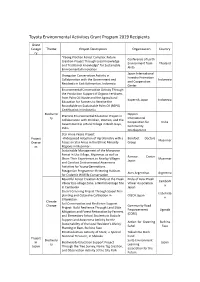
List of Previous Grant Projects
Toyota Environmental Activities Grant Program 2019 Recipients Grant Catego Theme Project Description Organization Country ry "Kaeng Krachan Forest Complex: Future Conference of Earth Creation Project Through Local Knowledge Environment from Thailand and Traditional Knowledge" for Sustainable Akita Environmental Innovation Japan International Orangutan Conservation Activity in Forestry Promotion Collaboration with the Government and Indonesia and Cooperation Residents in East Kalimantan, Indonesia Center Environmental Conservation Activity Through the Production Support of Organic Fertilizers from Palm Oil Waste and the Agricultural Kopernik Japan Indonesia Education for Farmers to Receive the Roundtable on Sustainable Palm Oil (RSPO) Certification in Indonesia Biodiversi Nippon Practical Environmental Education Project in ty International Collaboration with Children, Women, and the Cooperation for India Government in a Rural Village in Bodh Gaya, Community India Development Star Anise Peace Project Project -Widespread Adoption of Agroforestry with a Barefoot Doctors Myanmar Overse Focus on Star Anise in the Ethnic Minority Group as Regions in Myanmar- Sustainable Management of the Mangrove Forest in Uto Village, Myanmar, as well as Ramsar Center Share Their Experiences to Nearby Villages Myanmar Japan and Conduct Environmental Awareness Activities for Young Generations Patagonian Programme: Restoring Habitats Aves Argentinas Argentina for Endemic Wildlife Conservation Beautiful Forest Creation Activity at the Preah Pride of Asia: Preah -

Molecular Phylogenetics of the Genus Ceratitis (Diptera: Tephritidae)
Molecular Phylogenetics and Evolution 38 (2006) 216–230 www.elsevier.com/locate/ympev Molecular phylogenetics of the genus Ceratitis (Diptera: Tephritidae) Norman B. Barr ¤, Bruce A. McPheron Department of Entomology, Pennsylvania State University, University Park, PA 16802, USA Received 29 March 2005; revised 3 October 2005; accepted 5 October 2005 Abstract The Afrotropical fruit Xy genus Ceratitis MacLeay is an economically important group that comprises over 89 species, subdivided into six subgenera. Cladistic analyses of morphological and host use characters have produced several phylogenetic hypotheses for the genus. Only monophyly of the subgenera Pardalaspis and Ceratitis (sensu stricto) and polyphyly of the subgenus Ceratalaspis are common to all of these phylogenies. In this study, the hypotheses developed from morphological and host use characters are tested using gene trees pro- duced from DNA sequence data of two mitochondrial genes (cytochrome oxidase I and NADH-dehydrogenase subunit 6) and a nuclear gene (period). Comparison of gene trees indicates the following relationships: the subgenus Pardalaspis is monophyletic, subsection A of the subgenus Pterandrus is monophyletic, the subgenus Pterandrus may be either paraphyletic or polyphyletic, the subgenus Ceratalaspis is polyphyletic, and the subgenus Ceratitis s. s. might not be monophyletic. In addition, the genera Ceratitis and Trirhithrum do not form reciprocally monophyletic clades in the gene trees. Although the data statistically reject monophyly for Trirhithrum under the Shimoda- ira–Hasegawa test, they do not reject monophyly of Ceratitis. 2005 Elsevier Inc. All rights reserved. Keywords: Ceratitis; Trirhithrum; Tephritidae; ND6; COI; period 1. Introduction cies, C. capitata (Wiedemann) (commonly known as the Mediterranean fruit Xy), is already an invasive species The genus Ceratitis MacLeay (Diptera: Tephritidae) with established populations throughout tropical, sub- comprises over 89 Afrotropical species of fruit Xy (De tropical, and mild temperate habitats worldwide (Vera Meyer, 2000a).