3Rd EFMC Young Medicinal Chemist Symposium
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Fourth Report of Senior Pay and Perks in UK Universities History This
Transparency at the top? The fourth report of senior pay and perks in UK universities History This is the fourth report on pay and perks at the top of British higher education institutions (HEIs) to be published by the University and College Union (UCU). It forms part of the union’s ongoing campaign for greater transparency in higher education, including the rationale behind senior pay rises. UCU submitted a Freedom of Information (FoI) request to 158 HEIs in October 2017. This followed similar requests submitted in 2016, 2015 and 2014. All requests were designed to shine a light on the arbitrary nature of senior pay and perks in universities, and support the union’s call for reform. The basis for this report The FoI request that forms the basis of this report was sent to 158 (HEIs). It requested details of vice-chancellors’ (or head of institution if known by a different title) salaries and those of other senior post-holders earning over £100,000 at the institution during the academic year of 2016/17 (1 August 2016 to 31 July 2017). It also asked for details of flights, spending on hotels, spending on expenses and if the vice-chancellor was provided with accommodation by the university. Finally, we requested to know whether or not the vice-chancellor was a member of the remuneration committee, and requested a copy of the most recently ratified minutes of the institution’s remuneration committee. Variety of responses The questions on expenditure on flights, hotels, expenses and accommodation for vice-chancellors elicited a huge variation in responses with many institutions deploying exemptions under the Freedom of Information Act to avoid providing data. -
ANNNNNNNNNNNNNNNNNNNN 100A 006 Left Eye Input Right Eye Input
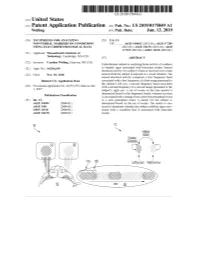
US 20190175049A1 ( 19) United States (12 ) Patent Application Publication (10 ) Pub. No. : US 2019 /0175049 A1 Welling ( 43 ) Pub . Date : Jun . 13 , 2019 ( 54 ) TECHNIQUES FOR ANALYZING (52 ) U . S . CI. NON -VERBAL MARKERS OF CONDITIONS CPC . .. A61B 5 /04842 (2013 . 01 ) ; A61B 5 / 7289 USING ELECTROPHYSIOLOGICAL DATA (2013 . 01) ; A61B 5 /0478 ( 2013 .01 ) ; A61B 5 /7225 ( 2013. 01 ) ; G06N 20 / 10 (2019 .01 ) (71 ) Applicant: Massachusetts Institute of Technology , Cambridge , MA (US ) ( 57 ) ABSTRACT (72 ) Inventor : Caroline Welling, Hanover, NH (US ) Embodiments related to analyzing brain activity of a subject to identify signs associated with binocular rivalry . Sensed ( 21 ) Appl. No. : 16 / 206, 639 electrical activity of a subject' s brain is received over a time period while the subject is exposed to a visual stimulus. The ( 22 ) Filed : Nov. 30 , 2018 sensed electrical activity comprises a first frequency band Related U . S . Application Data associated with a first frequency of a first image presented to the subject ' s left eye , a second frequency band associated (60 ) Provisional application No .62 / 593 , 535, filed on Dec . with a second frequency of a second image presented to the 1 , 2017 subject ' s right eye . A set of events in the time period is determined based on the frequency bands, wherein an event Publication Classification is associated with a change from a previous perceptual event (51 ) Int. Ci. to a new perceptual event. A metric for the subject is A61B 5 /0484 ( 2006 .01 ) determined based on the set of events . The metric is ana A61B 5 /00 ( 2006 .01 ) lyzed to determine whether the subject exhibits signs asso GO6N 20 / 10 (2006 .01 ) ciated with a condition that is associated with binocular A61B 5 /0478 ( 2006 .01 ) rivalry . -

Enthusing Children About Chemistry Climate Change and Biogenic Emissions Predicting Properties Using Informatics the Oil Industr
Summer 2008 Enthusing children about chemistry Predicting properties using informatics Climate change and biogenic emissions The oil industry’s chemical challenges As I see it... So are you finding it increasingly diffi - Oil exploration doesn’t just offer a career for engineers – cult to attract good chemists? chemists are vital, too. Sarah Houlton spoke to Schlumberger’s It can be a challenge, yes. Many of the com - pany’s chemists are recruited here, and they Tim Jones about the crucial role of chemistry in the industry often move on to other sites such as Houston or Paris, but finding them in the first place can be a challenge. Maybe one reason is that the oil People don’t think of Schlumberger as We can’t rely on being able to find suitable industry doesn’t have the greatest profile in a chemistry-using company, but an chemistries in other industries, either, mainly chemistry, and people think it employs engi - engineering one. How important is because of the high temperatures and pressures neers, not chemists. But it’s something the chemistry in oil exploration? that we have to be able to work at. Typically, the upstream oil industry cannot manage without, It’s essential! There are many challenges for upper temperature norm is now 175°C, but even if they don’t realise it! For me, maintaining chemistry in helping to maintain or increase oil we’re increasingly looking to go over 200°C. – if not enhancing – our recruitment is perhaps production. It’s going to become increasingly For heavy oil, where we heat the oil up with one of the biggest issues we face. -

Campro Catalog Stable Isotope
Introduction & Welcome Dear Valued Customer, We are pleased to present to you our Stable Isotopes Catalog which contains more than three thousand (3000) high quality labeled compounds. You will find new additions that are beneficial for your research. Campro Scientific is proud to work together with Isotec, Inc. for the distribution and marketing of their stable isotopes. We have been working with Isotec for more than twenty years and know that their products meet the highest standard. Campro Scientific was founded in 1981 and we provide services to some of the most prestigious universities, research institutes and laboratories throughout Europe. We are a research-oriented company specialized in supporting the requirements of the scientific community. We are the exclusive distributor of some of the world’s leading producers of research chemicals, radioisotopes, stable isotopes and environmental standards. We understand the requirements of our customers, and work every day to fulfill them. In working with us you are guaranteed to receive: - Excellent customer service - High quality products - Dependable service - Efficient distribution The highly educated staff at Campro’s headquarters and sales office is ready to assist you with your questions and product requirements. Feel free to call us at any time. Sincerely, Dr. Ahmad Rajabi General Manager 180/280 = unlabeled 185/285 = 15N labeled 181/281 = double labeled (13C+15N, 13C+D, 15N+18O etc.) 186/286 = 12C labeled 182/282 = d labeled 187/287 = 17O labeled 183/283 = 13C labeleld 188/288 = 18O labeled 184/284 = 16O labeled, 14N labeled 189/289 = Noble Gases Table of Contents Ordering Information.................................................................................................. page 4 - 5 Packaging Information .............................................................................................. -

Womencount: Leaders in Higher Education 2016
WomenCount Leaders in Higher Education 2016 A report by Norma Jarboe OBE ‘There’s no magic about getting a good gender balance – just dogged repetition of what a high priority it is and a determination to seek out strong women candidates who could hold their own against any competition.’ WomenCount ‘If universities inhibit the progression of talented female staff, they in turn are unable to reach their full potential. We know that universities make a huge contribution to society through research, teaching and partnerships with businesses, among many other activities.’ Professor Dame Athene Donald, Professor of Experimental Physics and Master of Churchill College, University of Cambridge WomenCount are very grateful to Perrett Laver for their support, the Government Equalities Office for their enagagement and Imperial College London for hosting the launch of this report on 2 March 2016. Cover quotation: Sir Nicholas Montagu, Chair of Council, Queen Mary University of London. Published by WomenCount © March 2016, all rights reserved. www.women-count.org Designed and produced by Graffeg. WomenCount: Leaders in Higher Education 2016 Contents 3 Foreword 4 Executive summary 5 Introduction 6 Collective action 9 Collegial governance: diversity challenge or opportunity? 10 Governing bodies: women’s representation on the rise 11 Governing bodies: the balancing act 12 Chairs: few seats for women 14 Vice-Chancellors: barely a fifth are women 15 Chair and Vice-Chancellor teams 16 Executive teams: a pipeline of women leaders 17 Academic heads: less than a third are women 19 HEI income impact women’s leadership 21 Mapping Women’s Leadership in HEIs 22 Reflections on the research 27 The Index 37 Biographies of new Chairs 42 Biographies of new Vice-Chancellors 47 About WomenCount and the author 1 Foreword ‘Gender equality is not a matter of being nice to women. -

Perspective Lecture, Frankfurt February 14Th 2013 Professor Paul
Professor Paul Workman Bio: Perspective Lecture, Frankfurt February 14 th 2013 Professor Paul Workman – Short Biography Professor Paul Workman is a leader in the discovery and development of molecularly targeted cancer drugs and is a passionate advocate of personalized cancer therapy. A molecular pharmacologist and chemical biologist, Paul has been responsible for the discovery and development of a large number of new molecularly targeted cancer drugs. Paul is currently Deputy Chief Executive of The Institute of Cancer Research (ICR) and Director of the ICR’s Cancer Research UK Cancer Therapeutics Unit in Sutton, UK, which is the largest non-profit cancer drug discovery group worldwide. He is also Head of ICR’s Division of Cancer Therapeutics and Harrap Professor of Pharmacology and Therapeutics. Paul obtained his BSc (Hons) in Biochemistry from Leicester University (1973) and his PhD in Cancer Pharmacology from Leeds University (1977). He then moved to Cambridge to become a postdoctoral fellow and scientific staff member of the MRC Clinical Oncology Unit, MRC Centre, Cambridge University (1976-1990) where he established and led the cancer pharmacology group, making major contributions to drugs targeting tumour hypoxia. Following a period as UICC Visiting Fellow at Stanford University and SRI International, California (1989), Paul was appointed as Cancer Research Campaign Professor and Director of Laboratory Research in the Department of Medical Oncology, Beatson Laboratories, Glasgow University (1990-1993). Paul then gained experience in the pharmaceutical industry. From 1993-1997 Paul was Head of the Cancer Research Bioscience Section at AstraZeneca Pharmaceuticals, Alderley Park, UK where he also initiated and led the strategic alliance with Sugen. -

Situación Actual Y Perspectivas De Futuro Personalized Precision Oncology: Current Status and Future Perspectives Madrid, 21 Y 22 De Marzo / March 21-22, 2019
Simposio Internacional International Symposium Oncología personalizada de precisión: situación actual y perspectivas de futuro Personalized Precision Oncology: current status and future perspectives Madrid, 21 y 22 de marzo / March 21-22, 2019 BIO Paul Workman Professor Paul Workman FMedSci, FRS is Chief Executive and President of The Institute of Cancer Research (ICR). Professor Workman is a passionate advocate of personalised molecular medicine and is an enthusiastic practitioner of multidisciplinary cancer drug discovery and development approaches to 'drugging the cancer genome'. He also conceptualised the 'Pharmacological Audit Trail' approach. As well as establishing and successfully leading drug discovery project teams yielding clinical candidates, Professor Workman’s personal and collaborative research utilises molecular pharmacology and chemical biology approaches, including high-throughput and genome-wide as well as hypothesis- driven strategies, to interrogate cancer biology, identify and validate new drug targets, discover and develop chemical tools and drugs acting on these targets, identify predictive and mechanism of action biomarkers, and elucidate mechanisms of drug sensitivity of resistance. He is especially interested in exploiting the addictions, vulnerabilities and dependencies of cancer cells using a combination of small molecule tools and drugs alongside molecular genetic techniques. Professor Workman has successfully built a series of multidisciplinary drug discovery and development teams in the academic, large pharma and biotechnology company sectors. Through this experience he has been able to combine the best elements of each of these environments. He has been responsible for the discovery of a number of drug development candidates, including in particular pathfinding inhibitors of the HSP90 molecular chaperone and the PI3 kinase family of signalling enzymes. -

5994392 Tion of Application No. 67375.734 Eb3-1685, PEN. T
USOO5994392A United States Patent (19) 11 Patent Number: 5,994,392 Shashoua (45) Date of Patent: Nov.30, 1999 54 ANTIPSYCHOTIC PRODRUGS COMPRISING 5,120,760 6/1992 Horrobin ................................. 514/458 AN ANTIPSYCHOTICAGENT COUPLED TO 5,141,958 8/1992 Crozier-Willi et al. ................ 514/558 AN UNSATURATED FATTY ACID 5,216,023 6/1993 Literati et al. .......................... 514/538 5,246,726 9/1993 Horrobin et al. ....................... 424/646 5,516,800 5/1996 Horrobin et al. ....................... 514/560 75 Inventor: Victor E. Shashoua, Brookline, Mass. 5,580,556 12/1996 Horrobin ................................ 424/85.4 73 Assignee: Neuromedica, Inc., Conshohocken, Pa. FOREIGN PATENT DOCUMENTS 30009 6/1981 European Pat. Off.. 21 Appl. No.: 08/462,820 009 1694 10/1983 European Pat. Off.. 22 Filed: Jun. 5, 1995 09 1694 10/1983 European Pat. Off.. 91694 10/1983 European Pat. Off.. Related U.S. Application Data 59-025327 2/1984 Japan. 1153629 6/1989 Japan. 63 Continuation of application No. 08/080,675, Jun. 21, 1993, 1203331 8/1989 Japan. abandoned, which is a continuation of application No. 07/952,191, Sep. 28, 1992, abandoned, which is a continu- (List continued on next page.) ation of application No. 07/577,329, Sep. 4, 1990, aban doned, which is a continuation-in-part of application No. OTHER PUBLICATIONS 07/535,812,tion of application Jun. 11, No. 1990, 67,375.734 abandoned, Eb3-1685, which is a continu-PEN. T. Higuchi et al. 66 Prodrugs as Noye Drug Delivery Sys 4,933,324, which is a continuation-in-part of application No. -
![Ecosystems for Innovation [Compatibility Mode]](https://docslib.b-cdn.net/cover/4930/ecosystems-for-innovation-compatibility-mode-1254930.webp)
Ecosystems for Innovation [Compatibility Mode]
Ecosystems for innovation Claire Ruskin CEO, Cambridge Network Innovation in Cambridge – how did it happen, how can we grow it and is it repeatable elsewhere? Material from Shai Vyakarnum – Judge Business School Tim Minshall – Institute for Manufacturing Claire Ruskin – CEO Cambridge Network Steven Ireland – East of England Inward Investment Cambridge Network – II EE www.cambridgenetwork.co.uk What’s special about Cambridge? The starting point … • A global ‘top five’ university: The University of Cambridge has 800 years at the top • Proximity to London and Europe : 5 international airports within 2 hours • Highly qualified employees: > 40% of people living in Cambridge having a high level qualification (compared to the national average of < 20%) • A few hi-tech companies back in the fifties • The start of a world class contract R&D cluster (the consultancies) from 1960 • => Evolving to a hi-tech cluster supporting > 143,000 jobs in the region. The cluster generates the equivalent of an NPV of £53bn in GDP. • => Good quality of life: Polls highlight Cambridgeshire as one of the best places to live in the UK • => Attitude: a good feeling about success and starting something new Cambridge Network – II EE www.cambridgenetwork.co.uk Why will Cambridge continue to have competitive advantage? • Diverse science base and research infrastructure, bringing excellent people to the Universities, business and medical organisations • Practice at innovation on demand as well as commercialisation • Collective learning and networking systems • Entrepreneurial -
The Chemistry Enterprise in 2015
The Chemistry Enterprise in 2015 William F. Carroll, Jr., Occidental Chemical Corp., ACS President 2005 Douglas J. Raber, GreenPoint Science ACKNOWLEDGMENT First, we would like to thank the members of the Senior Analysis. Thanks to those chemists who contributed specific Advisory Group who were interviewed extensively to com- predictions contained in the paper itself. Finally, thanks to all prise the first part of the project, the Situation Analysis. In the ACS members and staff who participated in or facilitated addition, we thank the Governance Advisory Team who dialog and debate; you provided the engine to bring the helped us with the strategy and peer review of the Situation Enterprise Project to completion. Senior Advisory Group Governance Advisory Team Samuel W. Bodman, Alvin L. Kwiram, Michael Betenbaugh, C. Dale Poulter, Deputy Secretary, U.S. University of Washington Johns Hopkins University University of Utah Department of the Treasury Robert S. Langer, Chris Hollinsed, Carolyn Ribes, Ronald Breslow, Massachusetts Institute of DuPont Co. Dow Chemical Co. Columbia University Technology Michael Jaffe, Ron Webb, Donald M. Burland, Jeffrey M. Lipton, New Jersey Institute of Procter & Gamble National Science Foundation Nova Chemicals Technology Marinda Wu, Ralph J. Cicerone, Thomas E. Reilly, Jr., Michael Nevill, Science is Fun! University of California, Irvine American Chemistry Council Solvay America, Inc. Thomas E. D’Ambra, Alfred P. Sattelberger, Albany Molecular Research, Inc. Los Alamos National Laboratory Predictions Peter B. Dervan, Jay Short, California Institute of Technology Diversa Corp. Ronald Breslow, Simon Campbell, Arthur B. Ellis, Jeffrey J. Siirola, Columbia University Royal Society of Chemistry National Science Foundation Eastman Chemical Co. -

BSCB Newsletter 2019A:BSCB Aut2k7
2019 BSCB Magazine BRITISH SOCIETY FOR CELL BIOLOGY 2019 CONTENTS BSCB Magazine News 2 Book reviews 8 Features 9 Meeting Reports 21 Summer students 25 Society Business 32 Editorial Front cover: microscopic Welcome to the 2019 BSCB Magazine! This year Mustafa Aydogan (University of Oxford), as well as to structure of pectoral fin and Susana and Stephen are filling in for our Newsletter BSCB postdoc poster of the year winners Dr Anna hypaxial muscles of a zebrafish Editor Ann Wheeler. We hope you will enjoy this Caballe (University of Oxford) and Dr Agata Gluszek- Danio rerio larvae at four days year’s magazine! Kustusz (University of Edinburgh). post fertilization. The immunostaining highlights the This year we had a number of fantastic one day In 2019, we will have our jointly BSCB-BSDB organization of fast (red) and meetings sponsored by BSCB. These focus meetings Spring meeting at Warwick University from 7th–10th slow (green) myosins. All nuclei are great way to meet and discuss your science with April, organised by BSCB members Susana Godinho are highlighted in blue (hoechst). experts in your field and to strengthen your network of and Vicky Sanz-Moreno. The programme for this collaborators within the UK. You can read more about meeting, which usually provides a broad spectrum of these meetings in the magazine. If you have an idea themes, has a focus on cancer biology: cell for a focus one day meeting, check how to apply for migration/invasion, organelle biogenesis, trafficking, funding on page 4. Our ambassadors have also been cell-cell communication. -

O'neill2020 Redacted.Pdf (7.617Mb)
This thesis has been submitted in fulfilment of the requirements for a postgraduate degree (e.g. PhD, MPhil, DClinPsychol) at the University of Edinburgh. Please note the following terms and conditions of use: This work is protected by copyright and other intellectual property rights, which are retained by the thesis author, unless otherwise stated. A copy can be downloaded for personal non-commercial research or study, without prior permission or charge. This thesis cannot be reproduced or quoted extensively from without first obtaining permission in writing from the author. The content must not be changed in any way or sold commercially in any format or medium without the formal permission of the author. When referring to this work, full bibliographic details including the author, title, awarding institution and date of the thesis must be given. Functional Characterisation of Spontaneously Active GABAA Receptors in Rat Dentate Gyrus Granule Cells Nathanael O’Neill B.Medsc. (Hons) Doctor of Philosophy The University of Edinburgh 2020 ii Abstract GABAA receptors (GABAARs) are the principal inhibitory neurotransmitter receptors in the adult mammalian central nervous system. GABAARs mediate two forms of inhibition: fast, phasic conductance; and slow, tonic conductance. Tonic conductance arises due to the persistent activation of GABAARs. This persistent activation can occur by GABA-dependent or GABA- independent mechanisms. Low concentrations of ambient GABA activate high affinity GABAARs located outside the synapse – at peri-/extra-synaptic sites – to generate GABA-dependent tonic conductance. In contrast, GABA-independent tonic conductance is generated by GABAARs that activate spontaneously, in the absence of GABA, due to constitutive receptor gating.