O'neill2020 Redacted.Pdf (7.617Mb)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
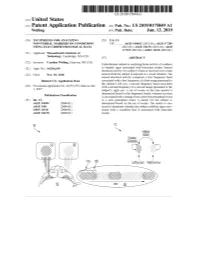
ANNNNNNNNNNNNNNNNNNNN 100A 006 Left Eye Input Right Eye Input
US 20190175049A1 ( 19) United States (12 ) Patent Application Publication (10 ) Pub. No. : US 2019 /0175049 A1 Welling ( 43 ) Pub . Date : Jun . 13 , 2019 ( 54 ) TECHNIQUES FOR ANALYZING (52 ) U . S . CI. NON -VERBAL MARKERS OF CONDITIONS CPC . .. A61B 5 /04842 (2013 . 01 ) ; A61B 5 / 7289 USING ELECTROPHYSIOLOGICAL DATA (2013 . 01) ; A61B 5 /0478 ( 2013 .01 ) ; A61B 5 /7225 ( 2013. 01 ) ; G06N 20 / 10 (2019 .01 ) (71 ) Applicant: Massachusetts Institute of Technology , Cambridge , MA (US ) ( 57 ) ABSTRACT (72 ) Inventor : Caroline Welling, Hanover, NH (US ) Embodiments related to analyzing brain activity of a subject to identify signs associated with binocular rivalry . Sensed ( 21 ) Appl. No. : 16 / 206, 639 electrical activity of a subject' s brain is received over a time period while the subject is exposed to a visual stimulus. The ( 22 ) Filed : Nov. 30 , 2018 sensed electrical activity comprises a first frequency band Related U . S . Application Data associated with a first frequency of a first image presented to the subject ' s left eye , a second frequency band associated (60 ) Provisional application No .62 / 593 , 535, filed on Dec . with a second frequency of a second image presented to the 1 , 2017 subject ' s right eye . A set of events in the time period is determined based on the frequency bands, wherein an event Publication Classification is associated with a change from a previous perceptual event (51 ) Int. Ci. to a new perceptual event. A metric for the subject is A61B 5 /0484 ( 2006 .01 ) determined based on the set of events . The metric is ana A61B 5 /00 ( 2006 .01 ) lyzed to determine whether the subject exhibits signs asso GO6N 20 / 10 (2006 .01 ) ciated with a condition that is associated with binocular A61B 5 /0478 ( 2006 .01 ) rivalry . -

Campro Catalog Stable Isotope
Introduction & Welcome Dear Valued Customer, We are pleased to present to you our Stable Isotopes Catalog which contains more than three thousand (3000) high quality labeled compounds. You will find new additions that are beneficial for your research. Campro Scientific is proud to work together with Isotec, Inc. for the distribution and marketing of their stable isotopes. We have been working with Isotec for more than twenty years and know that their products meet the highest standard. Campro Scientific was founded in 1981 and we provide services to some of the most prestigious universities, research institutes and laboratories throughout Europe. We are a research-oriented company specialized in supporting the requirements of the scientific community. We are the exclusive distributor of some of the world’s leading producers of research chemicals, radioisotopes, stable isotopes and environmental standards. We understand the requirements of our customers, and work every day to fulfill them. In working with us you are guaranteed to receive: - Excellent customer service - High quality products - Dependable service - Efficient distribution The highly educated staff at Campro’s headquarters and sales office is ready to assist you with your questions and product requirements. Feel free to call us at any time. Sincerely, Dr. Ahmad Rajabi General Manager 180/280 = unlabeled 185/285 = 15N labeled 181/281 = double labeled (13C+15N, 13C+D, 15N+18O etc.) 186/286 = 12C labeled 182/282 = d labeled 187/287 = 17O labeled 183/283 = 13C labeleld 188/288 = 18O labeled 184/284 = 16O labeled, 14N labeled 189/289 = Noble Gases Table of Contents Ordering Information.................................................................................................. page 4 - 5 Packaging Information .............................................................................................. -

Type of the Paper (Article
Supplementary Material A Proteomics Study on the Mechanism of Nutmeg-induced Hepatotoxicity Wei Xia 1, †, Zhipeng Cao 1, †, Xiaoyu Zhang 1 and Lina Gao 1,* 1 School of Forensic Medicine, China Medical University, Shenyang 110122, P. R. China; lessen- [email protected] (W.X.); [email protected] (Z.C.); [email protected] (X.Z.) † The authors contributed equally to this work. * Correspondence: [email protected] Figure S1. Table S1. Peptide fraction separation liquid chromatography elution gradient table. Time (min) Flow rate (mL/min) Mobile phase A (%) Mobile phase B (%) 0 1 97 3 10 1 95 5 30 1 80 20 48 1 60 40 50 1 50 50 53 1 30 70 54 1 0 100 1 Table 2. Liquid chromatography elution gradient table. Time (min) Flow rate (nL/min) Mobile phase A (%) Mobile phase B (%) 0 600 94 6 2 600 83 17 82 600 60 40 84 600 50 50 85 600 45 55 90 600 0 100 Table S3. The analysis parameter of Proteome Discoverer 2.2. Item Value Type of Quantification Reporter Quantification (TMT) Enzyme Trypsin Max.Missed Cleavage Sites 2 Precursor Mass Tolerance 10 ppm Fragment Mass Tolerance 0.02 Da Dynamic Modification Oxidation/+15.995 Da (M) and TMT /+229.163 Da (K,Y) N-Terminal Modification Acetyl/+42.011 Da (N-Terminal) and TMT /+229.163 Da (N-Terminal) Static Modification Carbamidomethyl/+57.021 Da (C) 2 Table S4. The DEPs between the low-dose group and the control group. Protein Gene Fold Change P value Trend mRNA H2-K1 0.380 0.010 down Glutamine synthetase 0.426 0.022 down Annexin Anxa6 0.447 0.032 down mRNA H2-D1 0.467 0.002 down Ribokinase Rbks 0.487 0.000 -

Compositions and Methods for Selective Delivery of Oligonucleotide Molecules to Specific Neuron Types
(19) TZZ ¥Z_T (11) EP 2 380 595 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 26.10.2011 Bulletin 2011/43 A61K 47/48 (2006.01) C12N 15/11 (2006.01) A61P 25/00 (2006.01) A61K 49/00 (2006.01) (2006.01) (21) Application number: 10382087.4 A61K 51/00 (22) Date of filing: 19.04.2010 (84) Designated Contracting States: • Alvarado Urbina, Gabriel AT BE BG CH CY CZ DE DK EE ES FI FR GB GR Nepean Ontario K2G 4Z1 (CA) HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL • Bortolozzi Biassoni, Analia Alejandra PT RO SE SI SK SM TR E-08036, Barcelona (ES) Designated Extension States: • Artigas Perez, Francesc AL BA ME RS E-08036, Barcelona (ES) • Vila Bover, Miquel (71) Applicant: Nlife Therapeutics S.L. 15006 La Coruna (ES) E-08035, Barcelona (ES) (72) Inventors: (74) Representative: ABG Patentes, S.L. • Montefeltro, Andrés Pablo Avenida de Burgos 16D E-08014, Barcelon (ES) Edificio Euromor 28036 Madrid (ES) (54) Compositions and methods for selective delivery of oligonucleotide molecules to specific neuron types (57) The invention provides a conjugate comprising nucleuc acid toi cell of interests and thus, for the treat- (i) a nucleic acid which is complementary to a target nu- ment of diseases which require a down-regulation of the cleic acid sequence and which expression prevents or protein encoded by the target nucleic acid as well as for reduces expression of the target nucleic acid and (ii) a the delivery of contrast agents to the cells for diagnostic selectivity agent which is capable of binding with high purposes. -

5994392 Tion of Application No. 67375.734 Eb3-1685, PEN. T
USOO5994392A United States Patent (19) 11 Patent Number: 5,994,392 Shashoua (45) Date of Patent: Nov.30, 1999 54 ANTIPSYCHOTIC PRODRUGS COMPRISING 5,120,760 6/1992 Horrobin ................................. 514/458 AN ANTIPSYCHOTICAGENT COUPLED TO 5,141,958 8/1992 Crozier-Willi et al. ................ 514/558 AN UNSATURATED FATTY ACID 5,216,023 6/1993 Literati et al. .......................... 514/538 5,246,726 9/1993 Horrobin et al. ....................... 424/646 5,516,800 5/1996 Horrobin et al. ....................... 514/560 75 Inventor: Victor E. Shashoua, Brookline, Mass. 5,580,556 12/1996 Horrobin ................................ 424/85.4 73 Assignee: Neuromedica, Inc., Conshohocken, Pa. FOREIGN PATENT DOCUMENTS 30009 6/1981 European Pat. Off.. 21 Appl. No.: 08/462,820 009 1694 10/1983 European Pat. Off.. 22 Filed: Jun. 5, 1995 09 1694 10/1983 European Pat. Off.. 91694 10/1983 European Pat. Off.. Related U.S. Application Data 59-025327 2/1984 Japan. 1153629 6/1989 Japan. 63 Continuation of application No. 08/080,675, Jun. 21, 1993, 1203331 8/1989 Japan. abandoned, which is a continuation of application No. 07/952,191, Sep. 28, 1992, abandoned, which is a continu- (List continued on next page.) ation of application No. 07/577,329, Sep. 4, 1990, aban doned, which is a continuation-in-part of application No. OTHER PUBLICATIONS 07/535,812,tion of application Jun. 11, No. 1990, 67,375.734 abandoned, Eb3-1685, which is a continu-PEN. T. Higuchi et al. 66 Prodrugs as Noye Drug Delivery Sys 4,933,324, which is a continuation-in-part of application No. -

Transport of Pregabalin Via L-Type Amino Acid Transporter 1 (SLC7A5) in Human Brain Capillary Endothelial Cell Line
Pharm Res (2018) 35: 246 https://doi.org/10.1007/s11095-018-2532-0 RESEARCH PAPER Transport of Pregabalin Via L-Type Amino Acid Transporter 1 (SLC7A5) in Human Brain Capillary Endothelial Cell Line Yu Takahashi1 & Tomohiro Nishimura 1 & Kei Higuchi 2 & Saki Noguchi 1 & Yuma Tega 2 & Toshiki Kurosawa2 & Yo s h i h a r u D e g u c h i 2 & Masatoshi Tomi1 Received: 14 July 2018 /Accepted: 21 October 2018 /Published online: 29 October 2018 # The Author(s) 2018 ABSTRACT Conclusions Our results indicate that LAT1, but not LAT2, Purpose The anti-epileptic drug pregabalin crosses the recognizes pregabalin as a substrate. It is suggested that LAT1 blood-brain barrier (BBB) in spite of its low lipophilicity. mediates pregabalin transport at the BBB. This study was performed to determine whether L-type amino acid transporters (LAT1/SLC7A5 and LAT2/SLC7A8) con- KEYWORDS anti-epileptic drug . blood-brain barrier . tribute to the uptake of pregabalin. L-type amino acid transporter . pregabalin Methods Pregabalin uptake by LATs-transfected HEK293 cells or hCMEC/D3 cells, an in vitro human BBB model, was measured by LC-MS/MS analysis. Expression of LAT1 mRNA in hCMEC/D3 cells was determined by quantitative ABBREVIATIONS RT-PCR analysis. BBB Blood-brain barrier Results Overexpression of LAT1, but not LAT2, in HEK293 BCH 2-Aminobicyclo-(2,2,1)-heptane-2-car- cells significantly increased the cellular uptake of pregabalin, boxylic acid GABA γ and the LAT1-mediated uptake was saturable with a Km of -Aminobutyric acid 0.288 mM. LAT1-mediated amino acid uptake was inhibited hCMEC/D3 cells Human immortalized brain endothelial specifically and almost completely in the presence of 1 mM cell line pregabalin. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

The Gabaa Receptor 8 Subunit Gene Promoter Fragments to Direct Long - Term Neuron - Specific Expression
THE GABAa RECEPTOR 8 SUBUNIT GENE PROMOTER : CHARACTERISATION AND USE Alexa Brett Roberts Division of Molecular Genetics Institute of Biomedical and Life Sciences University of Glasgow Glasgow A dissertation submitted for the degree of Doctor of Philosophy of the University of Glasgow February 1998 ProQuest Number: 13818612 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a com plete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest ProQuest 13818612 Published by ProQuest LLC(2018). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States C ode Microform Edition © ProQuest LLC. ProQuest LLC. 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106- 1346 GIASGOW UNIVERSITY LIBRARY 11110 (coh O f GLASGOW 1 IUNIVERSITT I [LQgAW I For my husband Stuart and for my children Ross and Ailie with love Also to JR (Grandad) in loving memory Preface The work presented in this thesis was performed entirely by the author, except where acknowledged. I declare that my thesis contains unique work and will not be submitted for any other degree, diploma or qualification at any other University. A. Brett Roberts December 1997 Acknowledgements I would like to thank Professor R.W. Davies for giving me the opportunity to study for this degree and for his help throughout the last three years. -

Tonic Gabaergic Inhibition As a New Way to Regulate Mesolimbic Dopamine System
Tonic GABAergic inhibition as a new way to regulate mesolimbic dopamine system Elena Vashchinkina Institute of Biomedicine, Pharmacology University of Helsinki Academic Dissertation To be presented with the permission of the Medical Faculty of the University of Helsinki, for public examination in lecture hall 3, Biomedicum Helsinki 1, Haartmaninkatu 8, on December 13th 2013 at 12 noon Helsinki 2013 Supervisor Professor Esa R. Korpi, MD PhD Institute of Biomedicine, Pharmacology Faculty of Medicine University of Helsinki, Finland Reviewers Docent Mikko Airavaara, PhD Institute of Biotechnology University of Helsinki, Finland Docent Tarja Stenberg, MD PhD Institute of Biomedicine, Physiology University of Helsinki, Finland Dissertation Opponent Professor Kimmo Jensen, MD PhD Department of Biomedicine Aarhus University, Denmark ISBN 978-952-10-9636-5 (paperback) ISBN 978-952-10-9637-2 (PDF) http://ethesis.helsinki.fi Unigrafia OY Helsinki 2013 TABLE OF CONTENTS Abstract ......................................................................................................................... 1 Original publications ................................................................................................... 2 Abbreviations ............................................................................................................... 3 Glossary of terms ......................................................................................................... 4 1 INTRODUCTION .................................................................................................... -

Neuroscience Products
Neuroscience Products CATALOG CATALOG NUMBER U.S. $ NUMBER U.S. $ -A- 3-(N-ACETYLAMINO)-5-(N-DECYL-N- 1 mg 27.50 159549 METHYLAMINO)BENZYL ALCOHOL 5 mg 89.40 o A23187 0-5 C [103955-90-4] (ADMB) See: Antibiotic A23187 A Protein Kinase C activator. Ref.: Proc. Nat. Acad. Sci. USA, 83, 4214 AA-861 20 mg 72.70 (1986). 159061 Purity: 95% 100 mg 326.40 C20H34N2O2 MW 334.5 0oC Orally active, specific and potent inhibitor of 5-lipoxygenase. N-ACETYL-ASP-GLU 25 mg 45.00 153036 [3106-85-2] 100 mg 156.00 Ref.: 1. Yoshimoto, T., et.al., Biochim. o Biophys. Acta, 713, 470 (1982). 2. Ashida, -20-0 C An endogenous neuropeptide with high 250 mg 303.65 Y., et.al., Prostaglandins, 26, 955 (1983). 3. affinity for a brain "Glutamate" receptor. Ancill, R.J., et.al., J. Int. Med. Res., 18, 75 Ref: Zaczek, R., et al., Proc. Natl. Acad. (1990). Sci. (USA), 80, 1116 (1983). C21H26O3 MW 326.4 C11H16N2O8 MW 304.3 ABL PROTEIN TYROSINE KINASE 250 U 47.25 N-ACETYL-2-BENZYLTRYPTAMINE 195876 (v-abl) 1 KU 162.75 See: Luzindole -70oC Recombinant Expressed in E. coli ACETYL-DL-CARNITINE 250 mg 60.00 A truncated form of the v-abl protein 154690 [2504-11-2] 1 g 214.00 tyrosine kinase which contains the 0oC Hydrochloride minimum region needed for kinase activity Crystalline and fibroblast transformation. Suppresses C9H17NO4 • HCl MW 239.7 apoptosis and induces resistance to anti-cancer compounds. O-ACETYL-L-CARNITINE CHLORIDE 500 mg 11.45 Activity: 100 KU/ml 159062 [5080-50-2] 1 g 20.65 Unit Definition: one unit is the amount of 0-5oC (R-(-)-2-Acetyloxy-3-carboxy-N,N,N-trimethyl 5 g 97.45 enzyme which catalyzes the transfer of 1 -1-propanaminium chloride) pmol of phosphate to EAIYAAPFAKKK per Purity: >88% minute at 30°C, pH 7.5. -
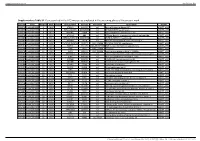
Supplementary Table S1. Genes Printed in the HC5 Microarray Employed in the Screening Phase of the Present Work
Supplementary material Ann Rheum Dis Supplementary Table S1. Genes printed in the HC5 microarray employed in the screening phase of the present work. CloneID Plate Position Well Length GeneSymbol GeneID Accession Description Vector 692672 HsxXG013989 2 B01 STK32A 202374 null serine/threonine kinase 32A pANT7_cGST 692675 HsxXG013989 3 C01 RPS10-NUDT3 100529239 null RPS10-NUDT3 readthrough pANT7_cGST 692678 HsxXG013989 4 D01 SPATA6L 55064 null spermatogenesis associated 6-like pANT7_cGST 692679 HsxXG013989 5 E01 ATP1A4 480 null ATPase, Na+/K+ transporting, alpha 4 polypeptide pANT7_cGST 692689 HsxXG013989 6 F01 ZNF816-ZNF321P 100529240 null ZNF816-ZNF321P readthrough pANT7_cGST 692691 HsxXG013989 7 G01 NKAIN1 79570 null Na+/K+ transporting ATPase interacting 1 pANT7_cGST 693155 HsxXG013989 8 H01 TNFSF12-TNFSF13 407977 NM_172089 TNFSF12-TNFSF13 readthrough pANT7_cGST 693161 HsxXG013989 9 A02 RAB12 201475 NM_001025300 RAB12, member RAS oncogene family pANT7_cGST 693169 HsxXG013989 10 B02 SYN1 6853 NM_133499 synapsin I pANT7_cGST 693176 HsxXG013989 11 C02 GJD3 125111 NM_152219 gap junction protein, delta 3, 31.9kDa pANT7_cGST 693181 HsxXG013989 12 D02 CHCHD10 400916 null coiled-coil-helix-coiled-coil-helix domain containing 10 pANT7_cGST 693184 HsxXG013989 13 E02 IDNK 414328 null idnK, gluconokinase homolog (E. coli) pANT7_cGST 693187 HsxXG013989 14 F02 LYPD6B 130576 null LY6/PLAUR domain containing 6B pANT7_cGST 693189 HsxXG013989 15 G02 C8orf86 389649 null chromosome 8 open reading frame 86 pANT7_cGST 693194 HsxXG013989 16 H02 CENPQ 55166 -

Structural and Functional Dynamics of Serotonin Transporter Gene Variants
STRUCTURAL AND FUNCTIONAL DYNAMICS OF SEROTONIN TRANSPORTER GENE VARIANTS By Meagan Anne Quinlan Dissertation Submitted to the Faculty of the Graduate School of Vanderbilt University In partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY in Pharmacology May 10, 2019 Nashville, Tennessee Approved: Brian Wadzinski, PhD Heidi Hamm, PhD Kevin Schey, PhD Randy Blakely, PhD ACKNOWLEDGEMENTS The completion of this dissertation would not be possible without the unwavering support of several people that have helped me immensely along the way. I first must give my greatest acknowledgment to my Ph.D. mentor Dr. Randy Blakely, without whom this accomplishment would not be possible. Being able to research the serotonin transporter, the protein he cloned back in 1989, has been the highest honor. His depth of knowledge and his unwavering support was unmatched during all the experiments I thought failed. He did always find that silver lining in the blank blots. The enthusiasm he gleaned when I would have exciting new data and even the countless hours we discussed the nuances of serotonin transporter kinetics as we trialed our way through data that didn’t always make sense, will definitely not be forgotten. Needless to say, there was not a better mentor that could have taken me through this journey. I was also lucky to have a supportive thesis committee. To Dr. Brain Wadzinski, Dr. Heidi Hamm, Dr. Ana Carnerio, and Dr. Kevin Schey, all this work definitely would have not been possible without all your insightful questions and constructive critiques. Thank you all for your encouragement and help over the past years.