Structural and Functional Dynamics of Serotonin Transporter Gene Variants
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Type of the Paper (Article
Supplementary Material A Proteomics Study on the Mechanism of Nutmeg-induced Hepatotoxicity Wei Xia 1, †, Zhipeng Cao 1, †, Xiaoyu Zhang 1 and Lina Gao 1,* 1 School of Forensic Medicine, China Medical University, Shenyang 110122, P. R. China; lessen- [email protected] (W.X.); [email protected] (Z.C.); [email protected] (X.Z.) † The authors contributed equally to this work. * Correspondence: [email protected] Figure S1. Table S1. Peptide fraction separation liquid chromatography elution gradient table. Time (min) Flow rate (mL/min) Mobile phase A (%) Mobile phase B (%) 0 1 97 3 10 1 95 5 30 1 80 20 48 1 60 40 50 1 50 50 53 1 30 70 54 1 0 100 1 Table 2. Liquid chromatography elution gradient table. Time (min) Flow rate (nL/min) Mobile phase A (%) Mobile phase B (%) 0 600 94 6 2 600 83 17 82 600 60 40 84 600 50 50 85 600 45 55 90 600 0 100 Table S3. The analysis parameter of Proteome Discoverer 2.2. Item Value Type of Quantification Reporter Quantification (TMT) Enzyme Trypsin Max.Missed Cleavage Sites 2 Precursor Mass Tolerance 10 ppm Fragment Mass Tolerance 0.02 Da Dynamic Modification Oxidation/+15.995 Da (M) and TMT /+229.163 Da (K,Y) N-Terminal Modification Acetyl/+42.011 Da (N-Terminal) and TMT /+229.163 Da (N-Terminal) Static Modification Carbamidomethyl/+57.021 Da (C) 2 Table S4. The DEPs between the low-dose group and the control group. Protein Gene Fold Change P value Trend mRNA H2-K1 0.380 0.010 down Glutamine synthetase 0.426 0.022 down Annexin Anxa6 0.447 0.032 down mRNA H2-D1 0.467 0.002 down Ribokinase Rbks 0.487 0.000 -

Compositions and Methods for Selective Delivery of Oligonucleotide Molecules to Specific Neuron Types
(19) TZZ ¥Z_T (11) EP 2 380 595 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 26.10.2011 Bulletin 2011/43 A61K 47/48 (2006.01) C12N 15/11 (2006.01) A61P 25/00 (2006.01) A61K 49/00 (2006.01) (2006.01) (21) Application number: 10382087.4 A61K 51/00 (22) Date of filing: 19.04.2010 (84) Designated Contracting States: • Alvarado Urbina, Gabriel AT BE BG CH CY CZ DE DK EE ES FI FR GB GR Nepean Ontario K2G 4Z1 (CA) HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL • Bortolozzi Biassoni, Analia Alejandra PT RO SE SI SK SM TR E-08036, Barcelona (ES) Designated Extension States: • Artigas Perez, Francesc AL BA ME RS E-08036, Barcelona (ES) • Vila Bover, Miquel (71) Applicant: Nlife Therapeutics S.L. 15006 La Coruna (ES) E-08035, Barcelona (ES) (72) Inventors: (74) Representative: ABG Patentes, S.L. • Montefeltro, Andrés Pablo Avenida de Burgos 16D E-08014, Barcelon (ES) Edificio Euromor 28036 Madrid (ES) (54) Compositions and methods for selective delivery of oligonucleotide molecules to specific neuron types (57) The invention provides a conjugate comprising nucleuc acid toi cell of interests and thus, for the treat- (i) a nucleic acid which is complementary to a target nu- ment of diseases which require a down-regulation of the cleic acid sequence and which expression prevents or protein encoded by the target nucleic acid as well as for reduces expression of the target nucleic acid and (ii) a the delivery of contrast agents to the cells for diagnostic selectivity agent which is capable of binding with high purposes. -

Transport of Pregabalin Via L-Type Amino Acid Transporter 1 (SLC7A5) in Human Brain Capillary Endothelial Cell Line
Pharm Res (2018) 35: 246 https://doi.org/10.1007/s11095-018-2532-0 RESEARCH PAPER Transport of Pregabalin Via L-Type Amino Acid Transporter 1 (SLC7A5) in Human Brain Capillary Endothelial Cell Line Yu Takahashi1 & Tomohiro Nishimura 1 & Kei Higuchi 2 & Saki Noguchi 1 & Yuma Tega 2 & Toshiki Kurosawa2 & Yo s h i h a r u D e g u c h i 2 & Masatoshi Tomi1 Received: 14 July 2018 /Accepted: 21 October 2018 /Published online: 29 October 2018 # The Author(s) 2018 ABSTRACT Conclusions Our results indicate that LAT1, but not LAT2, Purpose The anti-epileptic drug pregabalin crosses the recognizes pregabalin as a substrate. It is suggested that LAT1 blood-brain barrier (BBB) in spite of its low lipophilicity. mediates pregabalin transport at the BBB. This study was performed to determine whether L-type amino acid transporters (LAT1/SLC7A5 and LAT2/SLC7A8) con- KEYWORDS anti-epileptic drug . blood-brain barrier . tribute to the uptake of pregabalin. L-type amino acid transporter . pregabalin Methods Pregabalin uptake by LATs-transfected HEK293 cells or hCMEC/D3 cells, an in vitro human BBB model, was measured by LC-MS/MS analysis. Expression of LAT1 mRNA in hCMEC/D3 cells was determined by quantitative ABBREVIATIONS RT-PCR analysis. BBB Blood-brain barrier Results Overexpression of LAT1, but not LAT2, in HEK293 BCH 2-Aminobicyclo-(2,2,1)-heptane-2-car- cells significantly increased the cellular uptake of pregabalin, boxylic acid GABA γ and the LAT1-mediated uptake was saturable with a Km of -Aminobutyric acid 0.288 mM. LAT1-mediated amino acid uptake was inhibited hCMEC/D3 cells Human immortalized brain endothelial specifically and almost completely in the presence of 1 mM cell line pregabalin. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

O'neill2020 Redacted.Pdf (7.617Mb)
This thesis has been submitted in fulfilment of the requirements for a postgraduate degree (e.g. PhD, MPhil, DClinPsychol) at the University of Edinburgh. Please note the following terms and conditions of use: This work is protected by copyright and other intellectual property rights, which are retained by the thesis author, unless otherwise stated. A copy can be downloaded for personal non-commercial research or study, without prior permission or charge. This thesis cannot be reproduced or quoted extensively from without first obtaining permission in writing from the author. The content must not be changed in any way or sold commercially in any format or medium without the formal permission of the author. When referring to this work, full bibliographic details including the author, title, awarding institution and date of the thesis must be given. Functional Characterisation of Spontaneously Active GABAA Receptors in Rat Dentate Gyrus Granule Cells Nathanael O’Neill B.Medsc. (Hons) Doctor of Philosophy The University of Edinburgh 2020 ii Abstract GABAA receptors (GABAARs) are the principal inhibitory neurotransmitter receptors in the adult mammalian central nervous system. GABAARs mediate two forms of inhibition: fast, phasic conductance; and slow, tonic conductance. Tonic conductance arises due to the persistent activation of GABAARs. This persistent activation can occur by GABA-dependent or GABA- independent mechanisms. Low concentrations of ambient GABA activate high affinity GABAARs located outside the synapse – at peri-/extra-synaptic sites – to generate GABA-dependent tonic conductance. In contrast, GABA-independent tonic conductance is generated by GABAARs that activate spontaneously, in the absence of GABA, due to constitutive receptor gating. -
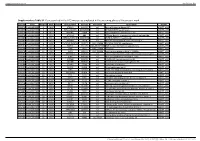
Supplementary Table S1. Genes Printed in the HC5 Microarray Employed in the Screening Phase of the Present Work
Supplementary material Ann Rheum Dis Supplementary Table S1. Genes printed in the HC5 microarray employed in the screening phase of the present work. CloneID Plate Position Well Length GeneSymbol GeneID Accession Description Vector 692672 HsxXG013989 2 B01 STK32A 202374 null serine/threonine kinase 32A pANT7_cGST 692675 HsxXG013989 3 C01 RPS10-NUDT3 100529239 null RPS10-NUDT3 readthrough pANT7_cGST 692678 HsxXG013989 4 D01 SPATA6L 55064 null spermatogenesis associated 6-like pANT7_cGST 692679 HsxXG013989 5 E01 ATP1A4 480 null ATPase, Na+/K+ transporting, alpha 4 polypeptide pANT7_cGST 692689 HsxXG013989 6 F01 ZNF816-ZNF321P 100529240 null ZNF816-ZNF321P readthrough pANT7_cGST 692691 HsxXG013989 7 G01 NKAIN1 79570 null Na+/K+ transporting ATPase interacting 1 pANT7_cGST 693155 HsxXG013989 8 H01 TNFSF12-TNFSF13 407977 NM_172089 TNFSF12-TNFSF13 readthrough pANT7_cGST 693161 HsxXG013989 9 A02 RAB12 201475 NM_001025300 RAB12, member RAS oncogene family pANT7_cGST 693169 HsxXG013989 10 B02 SYN1 6853 NM_133499 synapsin I pANT7_cGST 693176 HsxXG013989 11 C02 GJD3 125111 NM_152219 gap junction protein, delta 3, 31.9kDa pANT7_cGST 693181 HsxXG013989 12 D02 CHCHD10 400916 null coiled-coil-helix-coiled-coil-helix domain containing 10 pANT7_cGST 693184 HsxXG013989 13 E02 IDNK 414328 null idnK, gluconokinase homolog (E. coli) pANT7_cGST 693187 HsxXG013989 14 F02 LYPD6B 130576 null LY6/PLAUR domain containing 6B pANT7_cGST 693189 HsxXG013989 15 G02 C8orf86 389649 null chromosome 8 open reading frame 86 pANT7_cGST 693194 HsxXG013989 16 H02 CENPQ 55166 -

Embryonic Periventricular Endothelial Cells Demonstrate a Unique Pro
www.nature.com/scientificreports OPEN Embryonic periventricular endothelial cells demonstrate a unique pro‑neurodevelopment and anti‑infammatory gene signature Franciele Cristina Kipper1,5,7, Cleide Angolano2,5,7, Ravi Vissapragada1,4, Mauricio A. Contreras3, Justin Moore1,7, Manoj Bhasin6, Christiane Ferran2,3,5,7,8 & Ajith J. Thomas1,5,7,8* Brain embryonic periventricular endothelial cells (PVEC) crosstalk with neural progenitor cells (NPC) promoting mutual proliferation, formation of tubular‑like structures in the former and maintenance of stemness in the latter. To better characterize this interaction, we conducted a comparative transcriptome analysis of mouse PVEC vs. adult brain endothelial cells (ABEC) in mono‑culture or NPC co‑culture. We identifed > 6000 diferentially expressed genes (DEG), regardless of culture condition. PVEC exhibited a 30‑fold greater response to NPC than ABEC (411 vs. 13 DEG). Gene Ontology (GO) analysis of DEG that were higher or lower in PVEC vs. ABEC identifed “Nervous system development” and “Response to Stress” as the top signifcantly diferent biological process, respectively. Enrichment in canonical pathways included HIF1A, FGF/stemness, WNT signaling, interferon signaling and complement. Solute carriers (SLC) and ABC transporters represented an important subset of DEG, underscoring PVEC’s implication in blood–brain barrier formation and maintenance of nutrient‑rich/ non‑toxic environment. Our work characterizes the gene signature of PVEC and their important partnership with NPC, underpinning their unique role in maintaining a healthy neurovascular niche, and in supporting brain development. This information may pave the way for additional studies to explore their therapeutic potential in neuro‑degenerative diseases, such as Alzheimer’s and Parkinson’s disease. -

“The Transporter Transition”
Inaugural Conference “The Transporter Transition” September 18 – September 21, 2018 University Clinic of Dentistry Vienna Sensengasse 2a, 1090 Vienna, Austria We gratefully acknowledge the support of the following sponsors: 2 Organizers: ITTS Executive Committee: President: Harald Sitte (Medical University of Vienna) Vice-President (USA): Lynette Daws (University of Texas Health Science Center at San Antonio) Vice-President (EU): Balazs Sarkadi (Hungarian Academy of Sciences) Secretary/Treasurer: Habibeh Khoshbouei (University of Florida) Past President: Haley E. Melikian (University of Massachusetts Medical School) Administrative Assistant ITTS/SFB35, Local support: Daniela Prinz (Medical University of Vienna) Registration: University of Vienna Event Management and Conference Services of the University of Vienna Universitätsring 1 1010 Vienna 3 Table of Contents Scientific Program ................................................................................................................................... 5 Plenary Lectures .................................................................................................................................... 16 Session 1 ................................................................................................................................................ 20 Session 2 ................................................................................................................................................ 24 Session 3 ............................................................................................................................................... -

Phasic GABAA-Mediated Inhibition
Jasper's Basic Mechanisms of the Epilepsies Jasper's Basic Mechanisms of the Epilepsies Phasic GABAA-Mediated Inhibition Enrico Cherubini Neurobiology Department, International School for Advanced Studies (SISSA), Via Bonomea 265, 34136 Trieste, Italy Abstract Phasic inhibition consists of fast GABAA-mediated inhibitory postsynaptic potentials (IPSPs) regulating point to point communication between neurons. In physiological conditions it exerts a powerful control on cell excitability and network oscillations thought to be associated with high cognitive functions. GABAA receptors facing the presynaptic release sites are exposed, for a brief period of time, to high concentration of GABA released by exocytosis of presynaptic vesicles. Once released, GABA diffuses throughout the neuropil before being taken up by selective plasma membrane transporters which contribute to its clearance. In this Chapter different steps contributing to shape synaptic currents, including vesicle exocytosis, dynamics of GABA transient in the synaptic cleft, postsynaptic receptors and GABA transporters will be highlighted. Particular attention will be paid to the regulation of GABA release by the calcium-induced calcium release process and by presynaptic GABAA and/or GABAB receptors. Finally, the heterogeneity of GABAA receptor subunits will be discussed in relation to their ability to give rise to a variety of functional GABAA receptors, differently regulated by endogenous or exogenous molecules useful for the treatment of severe neurological and psychiatric disorders. γ-Aminobutiric acid (GABA) is the main inhibitory neurotransmitter in the adult mammalian CNS. It inhibits neuronal firing by activating two different classes of receptors, GABAA and GABAB. Unlike GABAA receptors which form integral ion channels, GABAB receptors are coupled to ion channels via guanine nucleotide-binding proteins and second messengers. -

The Role of Amino Acids in Neurotransmission and Fluorescent Tools for Their Detection
International Journal of Molecular Sciences Review The Role of Amino Acids in Neurotransmission and Fluorescent Tools for Their Detection Rochelin Dalangin 1, Anna Kim 1 and Robert E. Campbell 1,2,* 1 Department of Chemistry, University of Alberta, Edmonton, AB T6G 2G2, Canada; [email protected] (R.D.); [email protected] (A.K.) 2 Department of Chemistry, Graduate School of Science, The University of Tokyo, Bunkyo City, Tokyo 113-0033, Japan * Correspondence: [email protected]; Tel.: +1-7804921849 Received: 29 July 2020; Accepted: 24 August 2020; Published: 27 August 2020 Abstract: Neurotransmission between neurons, which can occur over the span of a few milliseconds, relies on the controlled release of small molecule neurotransmitters, many of which are amino acids. Fluorescence imaging provides the necessary speed to follow these events and has emerged as a powerful technique for investigating neurotransmission. In this review, we highlight some of the roles of the 20 canonical amino acids, GABA and β-alanine in neurotransmission. We also discuss available fluorescence-based probes for amino acids that have been shown to be compatible for live cell imaging, namely those based on synthetic dyes, nanostructures (quantum dots and nanotubes), and genetically encoded components. We aim to provide tool developers with information that may guide future engineering efforts and tool users with information regarding existing indicators to facilitate studies of amino acid dynamics. Keywords: amino acids; neurotransmission; fluorescence; imaging; biosensors; neurotransmitters; indicators 1. Introduction Neurons communicate to each other by the release of chemicals stored in synaptic vesicles across specialized gaps known as synapses. These chemicals diffuse across the synapse and bind to their target receptors on adjacent neurons to modulate their physiological states. -

Zinc in the Retinal Pigment Epithelium and Choriocapillaris Interface
Zinc in the Retinal Pigment Epithelium and Choriocapillaris Interface A Thesis Submitted to University College London for the degree of Doctor of Philosophy Sabrina Cahyadi BSc Department of Ocular Biology and Therapeutics Institute of Ophthalmology University College London 2012 1 Declaration I, Sabrina Cahyadi confirm that the work presented in the thesis titled “Zinc in the Retinal Pigment Epithelium and Choriocapillaris Interface” is my own work. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. ______________ Sabrina Cahyadi Date: 14 November 2011 2 Acknowledgements “For wisdom will enter your heart and knowledge will be pleasant to your soul; discretion will guard you, understanding will watch over you.” First and foremost, I would like to thank my supervisors Professor Phil Luthert and Dr. Imre Lengyel for accepting me as their student, and the enormous support, help and guidance they have provided. I would also like to thank the Dorothy Hodgkin Postgraduate Award, the Mercer Fund and Professor Alan Bird for their very generous studentship which enabled me to do this PhD. The funding I receive from the Special Trustees of Moorfields Eye Hospital allowed me to do my day-to-day lab works. I thank Peter Marshall who ensured that the funds reach me in time. I must also thank Dr. Peter Munro, Cynthia Langley, Alexandra Boss, and Dr. Virginia Calder for all the help and moral supports I have received. I cannot believe how much have happened in the past three years. The people at the Institute of Ophthalmology have supported me through all the amazing, good, bad, ho-hum, and heartbreaking episodes of my PhD. -

Supplemental Figures 04 12 2017
Jung et al. 1 SUPPLEMENTAL FIGURES 2 3 Supplemental Figure 1. Clinical relevance of natural product methyltransferases (NPMTs) in brain disorders. (A) 4 Table summarizing characteristics of 11 NPMTs using data derived from the TCGA GBM and Rembrandt datasets for 5 relative expression levels and survival. In addition, published studies of the 11 NPMTs are summarized. (B) The 1 Jung et al. 6 expression levels of 10 NPMTs in glioblastoma versus non‐tumor brain are displayed in a heatmap, ranked by 7 significance and expression levels. *, p<0.05; **, p<0.01; ***, p<0.001. 8 2 Jung et al. 9 10 Supplemental Figure 2. Anatomical distribution of methyltransferase and metabolic signatures within 11 glioblastomas. The Ivy GAP dataset was downloaded and interrogated by histological structure for NNMT, NAMPT, 12 DNMT mRNA expression and selected gene expression signatures. The results are displayed on a heatmap. The 13 sample size of each histological region as indicated on the figure. 14 3 Jung et al. 15 16 Supplemental Figure 3. Altered expression of nicotinamide and nicotinate metabolism‐related enzymes in 17 glioblastoma. (A) Heatmap (fold change of expression) of whole 25 enzymes in the KEGG nicotinate and 18 nicotinamide metabolism gene set were analyzed in indicated glioblastoma expression datasets with Oncomine. 4 Jung et al. 19 Color bar intensity indicates percentile of fold change in glioblastoma relative to normal brain. (B) Nicotinamide and 20 nicotinate and methionine salvage pathways are displayed with the relative expression levels in glioblastoma 21 specimens in the TCGA GBM dataset indicated. 22 5 Jung et al. 23 24 Supplementary Figure 4.